

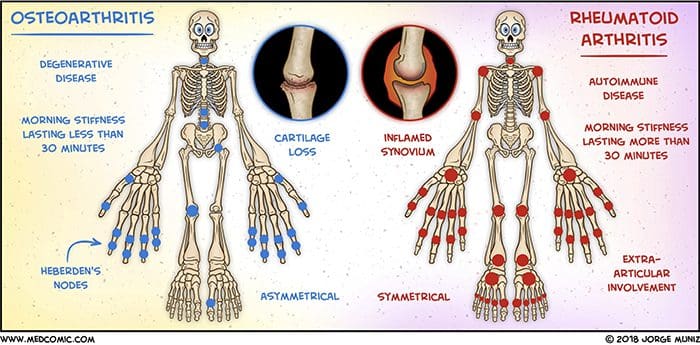
El Paso, TX. Chiropractor Dr. Alexander Jimenez takes a look at various conditions that can cause chronic pain. These include:
- Osteoarthritis
- Facetogenic pain
- Neuropathic pain
- HeadachesÂ

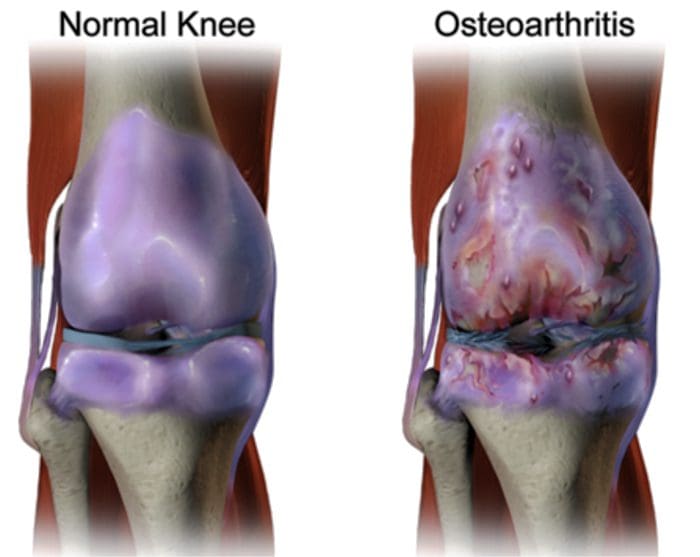
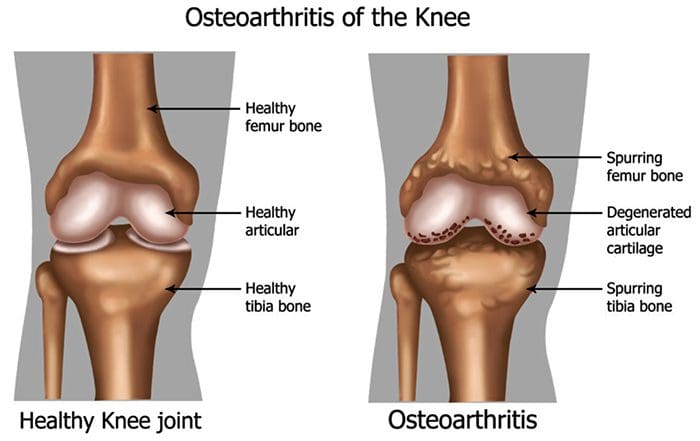
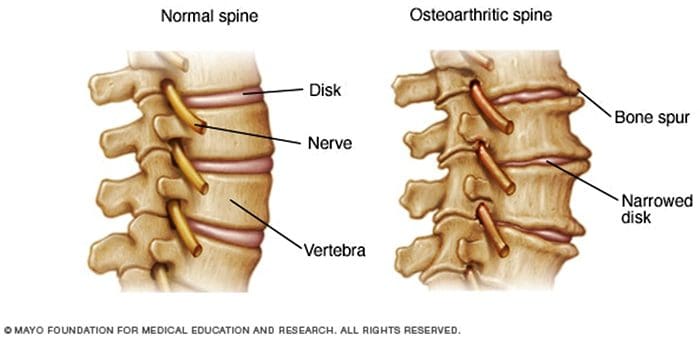
Table of Contents
 Abstract
Abstract
 Abstract
AbstractArthritis pain is a complex phenomenon involving intricate neurophysiological processing at all levels of the pain pathway. The treatment options available to alleviate joint pain are fairly limited and most arthritis patients report only modest pain relief with current treatments. A better understanding of the neural mechanisms responsible for musculoskeletal pain and the identification of new targets will help in the development of future pharmacological therapies. This article reviews some of the latest research into factors which contribute to joint pain and covers areas such as cannabinoids, proteinase activated receptors, sodium channels, cytokines and transient receptor potential channels. The emerging hypothesis that osteoarthritis may have a neuropathic component is also discussed.
Introduction
The world health organization ranks musculoskeletal disorders as the most frequent cause of disability in the modern world, affecting one in three adults [1]. Even more alarming is that the prevalence of these diseases is rising while our knowledge of their underlying causes is fairly rudimentary.
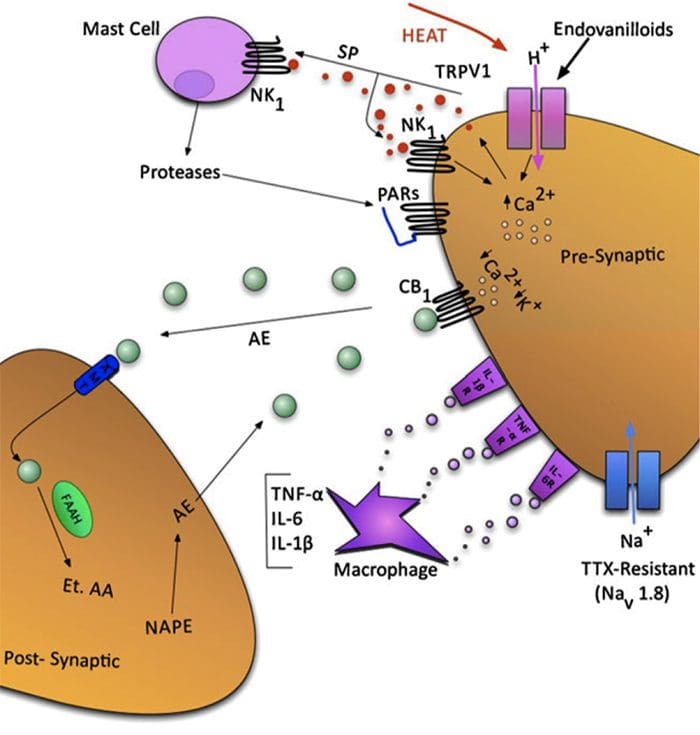
Fig. 1 A schematic illustrating some of the targets known to modulate joint pain. Neuromodulators can be released from nerve terminals as well as mast cells and macrophages to alter afferent mechanosensitivity. Endovanilloids, acid and noxious heat can activate transient receptor potential vanilloid type 1 (TRPV1) ion channels leading to the release of algogenic substance P (SP) which subsequently binds toneurokinin-1 (NK1) receptors. Proteases can cleave and stimulate pro-tease activated receptors (PARs). Thus far, PAR2and PAR4have been shown to sensitize joint primary afferents. The endocannabinoid anandamide (AE) is produced on demand and synthesized from N-arachidonylphosphatidylethanolamine (NAPE) under the enzymaticaction of phospholipases. A portion of AE then binds to cannabinoid-1 (CB1) receptors leading to neuronal desensitization. Unbound AE is rapidly taken up by an anandamide membrane transporter (AMT)before being broken down by a fatty acid amide hydrolase (FAAH)into ethanolamine (Et) and arachidonic acid (AA). The cytokines tumour necrosis factor-α(TNF-α), interleukin-6 (IL-6) and interleukin1-beta (IL-1β) can bind to their respective receptors to enhance pain transmission. Finally, there are tetrodotoxin (TTX)-resistant sodium channels (Nav1.8) involved in neuronal sensitization.
Patients yearn for their chronic pain to disappear; however, currently-prescribed analgesics are largely ineffective and are accompanied by a wide range of unwanted side effects.As such, millions of people worldwide are suffering from the debilitating effects of joint pain, for which there is no satisfactory treatment [2].
There are more than 100 different forms of arthritis with osteoarthritis (OA) being the most common. OA is a progressively degenerative joint disease that causes chronic pain and loss of function. Commonly, OA is the inability of the joint to repair damage effectively in response to excessive forces being placed on it. The biological and psychosocial factors that comprise chronic OA pain are not well understood, al-though ongoing research continues to unravel the complex nature of disease symptoms [2]. Current therapeutics, such as non-steroidal anti-inflammatory drugs (NSAIDs), provide some symptomatic relief, reducing the pain for short periods of time, but do not alleviate pain across the lifespan of the patient. Furthermore, high-dose NSAIDs cannot be taken repeatedly over many years, as this can lead to renal toxicity and gastrointestinal bleeding.
Traditionally, arthritis research has focused largely on the articular cartilage as a primary target for therapeutic development of novel OA drugs for disease modification. This“chondrocentric†focus has shed new light on the intricate biochemical and biomechanical factors that influence chondrocyte behavior in diseased joints. However, as articular cartilage is aneural and avascular, this tissue is unlikely to be the source of OA pain. This fact, coupled with the findings that there is no correlation between the damage of articular cartilage and pain in OA patients [3,4], or in preclinical models of OA [5••], has caused a shift in focus to develop drugs for effective pain control. This article will review the latest findings in joint pain research and highlight some of the emerging targets that may be the future of arthritis pain management (summarized in Fig. 1)
Cytokines
The actions of various cytokines in joint neurophysiology studies have featured quite prominently recently. Interleukin-6 (IL-6), for example, is a cytokine that typically binds to the membrane-bound IL-6 receptor (IL-6R). IL-6 can also signal by binding with a soluble IL-6R (sIL-6R) to produce an IL-6/sIL-6R complex. This IL-6/sIL-6R complex subsequent lybinds to a transmembrane glycoprotein subunit 130(gp130) thereby allowing IL-6 to signal in cells that do not constitutively express membrane-bound IL-6R [25,26]. IL-6 and sIL-6R are known to be key players in systemic inflammation and arthritis, as up regulation of both has been found in the serum and synovial fluid of RA patients [27–29]. Recently, Vazquez et al.observed that co-administration of IL-6/sIL-6R into rat knees caused inflammation-evoked pain, as revealed by an increase in the response of spinal dorsal horn neurons to mechanical stimulation of the knee and other parts of the hindlimb [30].Spinal neurone hyperexcitability was also seen when IL-6/sIL-6R was applied locally to the spinal cord. Spinal application of soluble gp130 (which would “mop upâ€IL-6/sIL-6R complexes thereby reducing trans signaling) inhibited IL-6/sIL-6R-induced central sensitization. Acute application of soluble gp130 alone, however, did not reduce the neuronal responses to already established joint inflammation.
The transient receptor potential (TRP) channels are a group of non-selective cation channels that act as integrators of various physiological and pathophysiological processes. In addition to thermosensation, chemosensation and mechanosensation, TRP channels are involved in the modulation of pain and inflammation. TRP vanilloid-1 (TRPV1) ion channels have been shown to contribute to joint inflammatory pain as thermal hyperalgesia was not evocable in TRPV1−/−monoarthritic mice [31]. Similarly, TRP ankyrin-1 (TRPA1)ion channels are involved in arthritic mechanohypersensitivity as blockade of the receptor with selective antagonistsattenuated mechanical pain in the Freund’s complete adjuvant model of inflammation [32,33]. Further evidence thatTRPV1 may be involved in the neurotransmission of OA pain comes from studies in which neuronal TRPV1 expression is elevated in the sodium monoiodoacetate model of OA [34]. Inaddition, systemic administration of the TRPV1 antagonist A-889425 reduced the evoked and spontaneous activity ofspinal-wide dynamic range and nociception-specific neuronesin the monoiodoacetate model [35]. These data suggest that endovanilloids could be involved in central sensitization processes associated with OA pain.
There are currently known to be at least four polymorphisms in the gene that encodes TRPV1, which can lead toan alteration in the structure of the ion channel and impaired function. One particular polymorphism (rs8065080) alters the sensitivity of TRPV1 to capsaicin and individuals carrying this polymorphism are less sensitive to thermal hyperalgesia [36]. Based on this genetic anomaly, a recent study examined whether OA patients with the rs8065080 poly-morphism experienced altered pain perception. The research team found that patients with asymptomatic knee OA were more likely to carry the rs8065080 gene than patients with painful joints [37]. This observation indicates that OA patients with normal functioning TRPV1 channels have an increased risk of joint pain and re-affirms the potential involvement of TRPV1 in OA pain perception.
Conclusion
While the hurdle of treating arthritis pain effectively still remains, great leaps are being made in our understanding of the neurophysiological processes responsible for the generation of joint pain. New targets are being discovered continually, while the mechanisms behind known pathways are being further defined and refined. Targeting one specific receptor or ion channel is unlikely to be the solution to normalizing joint pain, but rather a polypharmacy approach is indicated in which various mediators are used in combination during specific phases of the disease. Unravelling the functional circuitry at each level of the pain pathway will also improve our knowledge of how joint pain is generated. For example, identifying the peripheral mediators of joint pain will allow us to control nociception within the joint itself and likely avoid the central side effects of systemically-administered pharamcotherapeutics.
FACETOGENIC PAIN
FACET SYNDROME & FACETOGENIC PAIN
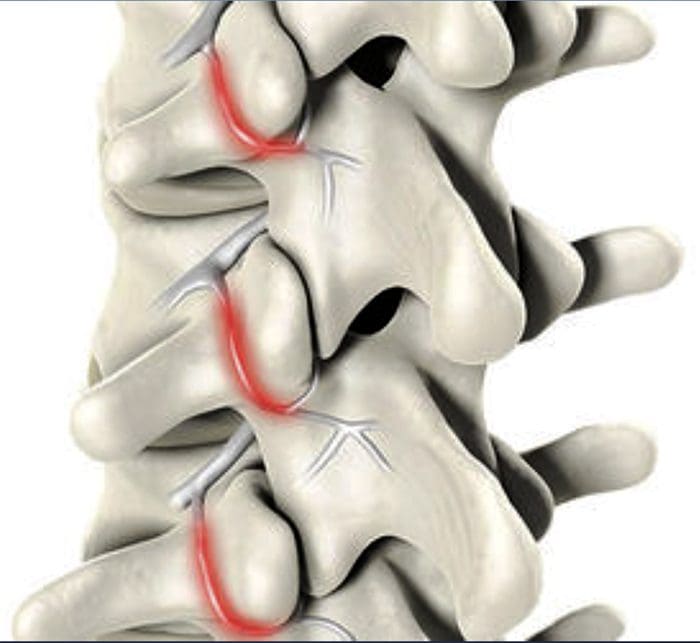 FACET SYNDROME & FACETOGENIC PAIN
FACET SYNDROME & FACETOGENIC PAIN- Facet syndrome is an articular disorder related to the lumbar facet joints and their innervations, and produces both local and radiating facetogenic pain.
- Excessive rotation, extension, or flexion of the spine (repeated overuse) can result in degenerative changes to the cartilage of the joint and may involve degenerative changes to other structures including the intervertebral disc
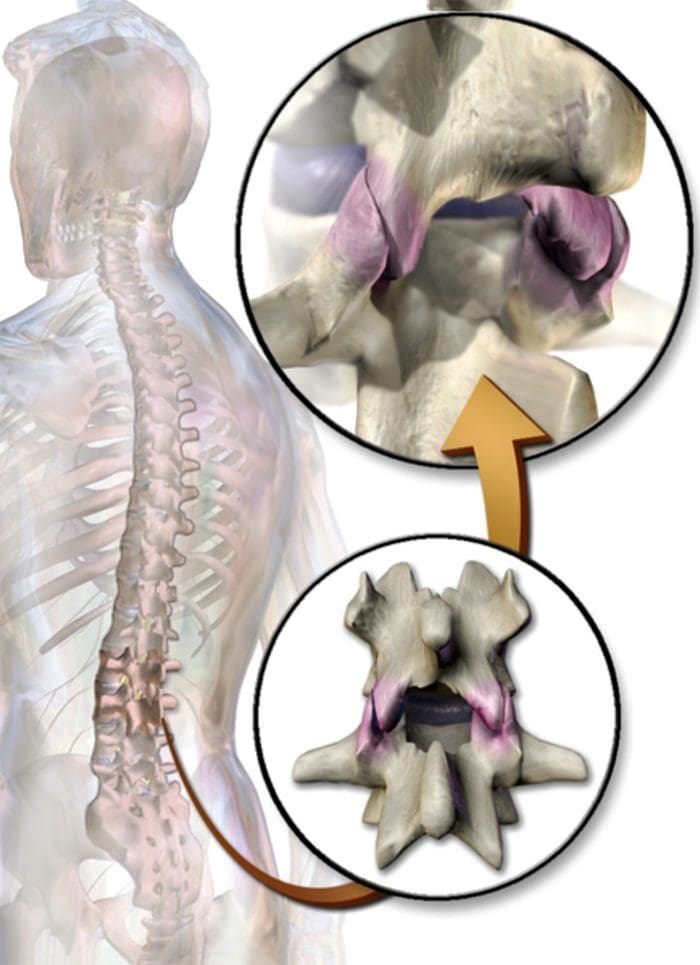
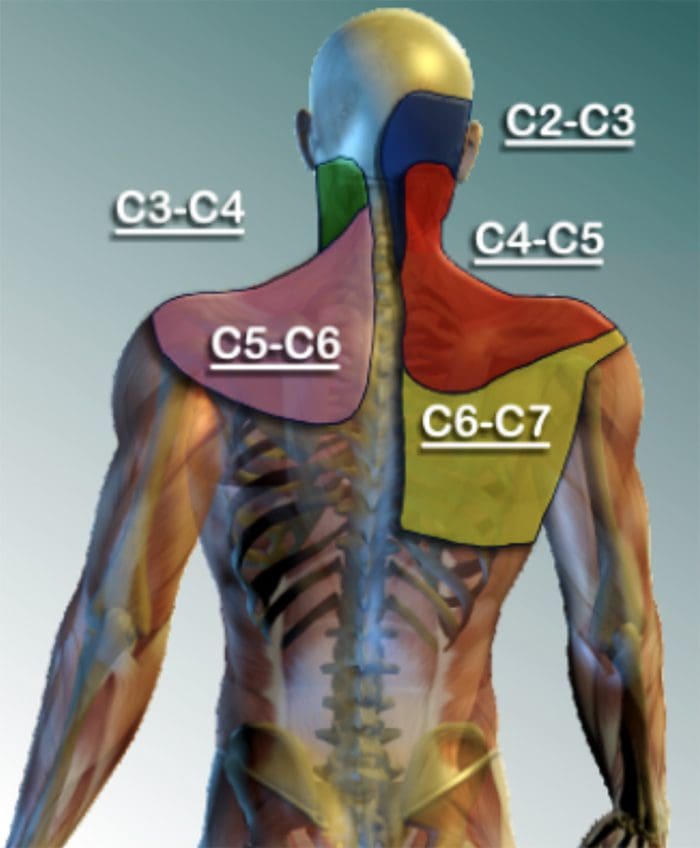
CERVICAL FACET SYNDROME & FACETOGENIC PAIN
- Axial neck pain (rarely radiating past the shoulders), most common unilaterally
- Pain with and/or limitation of extension and rotation
- Tenderness upon palpation
- Radiating facetogenic pain locally or into the shoulders or upper back, and rarely radiate in the front or down an arm or into the fingers as a herniated disc might.
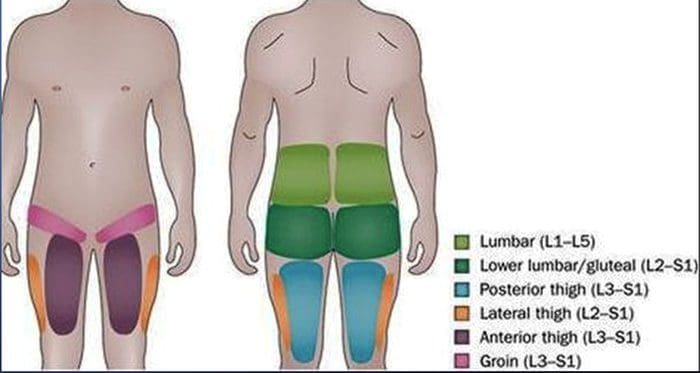
LUMBAR FACET SYNDROME & FACETOGENIC PAIN
 LUMBAR FACET SYNDROME & FACETOGENIC PAIN
LUMBAR FACET SYNDROME & FACETOGENIC PAIN- Pain or tenderness in lower back.
- Local tenderness/stiffness alongside the spine in the lower back.
- Pain, stiffness or difficulty with certain movements (such as standing up straight or getting up from a chair.
- Pain upon hyperextension
- Referred pain from upper lumbar facet joints can extend into the flank, hip and upper lateral thigh
- Referred pain from lower lumbar facet joints can penetrate deep into the thigh, laterally and/or posteriorly
- L4-L5 and L5-S1 facet joints can refer pain extending into the distal lateral leg, and in rare instances to the foot
EVIDENCE-BASED MEDICINE
 EVIDENCE-BASED MEDICINE
EVIDENCE-BASED MEDICINEEvidence-based Interventional Pain Medicine according to Clinical Diagnoses
12. Pain Originating from the Lumbar Facet Joints
Abstract
Although the existence of a “facet syndrome†had long been questioned, it is now generally accepted as a clinical entity. Depending on the diagnostic criteria, the zygapophysial joints account for between 5% and 15% of cases of chronic, axial low back pain. Most commonly, facetogenic pain is the result of repetitive stress and/or cumulative low level trauma, leading to inflammation and stretching of the joint capsule. The most frequent complaint is axial low back pain with referred pain perceived in the flank, hip, and thigh. No physical examination findings are pathognomonic for diagnosis. The strongest indicator for lumbar facetogenic pain is pain reduction after anesthetic blocks of the rami mediales (medial branches) of the rami dorsales that innervate the facet joints. Because false-positive and, possibly, false negative results may occur, results must be interpreted carefully. In patients with injection-confirmed zygapophysial joint pain, procedural interventions can be undertaken in the context of a multidisciplinary, multimodal treatment regimen that includes pharmacotherapy, physical therapy and regular exercise, and, if indicated, psychotherapy. Currently, the “gold standard†for treating facetogenic pain is radio frequency treatment (1 B+). The evidence supporting intra-articular corticosteroids is limited; hence, this should be reserved for those individuals who do not respond to radio frequency treatment (2 B1).
Facetogenic Pain emanating from the lumbar facet joints is a common cause of low back pain in the adult population. Golthwaite was the first to describe the syndrome in 1911, and Ghormley is generally credited with coining the term “facet syndrome†in 1933. Facetogenic pain is defined as pain that arises from any structure that is part of the facet joints, including the fibrous capsule, synovial membrane, hyaline cartilage, and bone.3–5
More commonly, it is the result of repetitive stress and/or cumulative low-level trauma. This leads to inflammation, which can cause the facet joint to be filled with fluid and swell, which in turn results in stretching of the joint capsule and subsequent pain generation.27 Inflammatory changes around the facet joint can also irritate the spinal nerve via foraminal narrowing, resulting in sciatica. In addition, Igarashi et al.28 found that inflammatory cytokines released through the ventral joint capsule in patients with zygapophysial joint degeneration may be partially responsible for the neuropathic symptoms in individuals with spinal stenosis. Predisposing factors for zygapophysial joint pain include spondylolisthesis/lysis, degenerative disc disease, and advanced age.5
I.C ADDITIONAL TESTS
The prevalence rate of pathological changes in the facet joints on radiological examination depends on the mean age of the subjects, the radiological technique used, and the definition of “abnormality.†Degenerative facet joints can be best visualized via computed tomography (CT) examination.49
NEUROPATHIC PAIN

- Pain initiated or caused by a primary lesion or dysfunction in the somatosensory nervous system.
- Neuropathic pain is usually chronic, difficult to treat and often resistant to standard analgesic management.
Abstract
Neuropathic pain is caused by a lesion or disease of the somatosensory system, including peripheral fibres (Aβ, Aδ and C fibres) and central neurons, and affects 7–10% of the general population. Multiple causes of neuropathic pain have been described and its incidence is likely to increase owing to the ageing global population, increased incidence of diabetes mellitus and improved survival from cancer after chemotherapy. Indeed, imbalances between excitatory and inhibitory somatosensory signalling, alterations in ion channels and variability in the way that pain messages are modulated in the central nervous system all have been implicated in neuropathic pain. The burden of chronic neuropathic pain seems to be related to the complexity of neuropathic symptoms, poor outcomes and difficult treatment decisions. Importantly, quality of life is impaired in patients with neuropathic pain owing to increased drug prescriptions and visits to health care providers, as well as the morbidity from the pain itself and the inciting disease. Despite challenges, progress in the understanding of the pathophysiology of neuropathic pain is spurring the development of new diagnostic procedures and personalized interventions, which emphasize the need for a multidisciplinary approach to the management of neuropathic pain.
PATHOGENESIS OF NEUROPATHIC PAIN
-
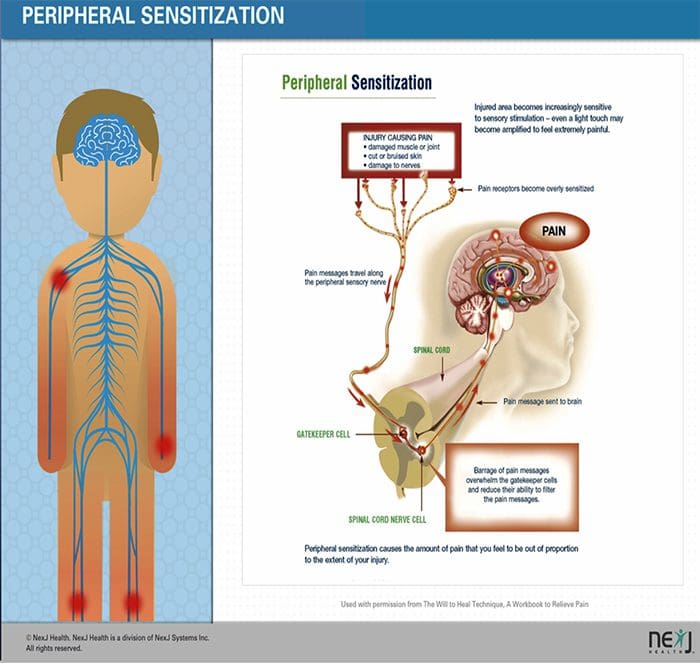
PERIPHERAL MECHANISMS
- After a peripheral nerve lesion, neurons become more sensitive and develop abnormal excitability and elevated sensitivity to stimulation
- This is known as…Peripheral Sensitization!
-
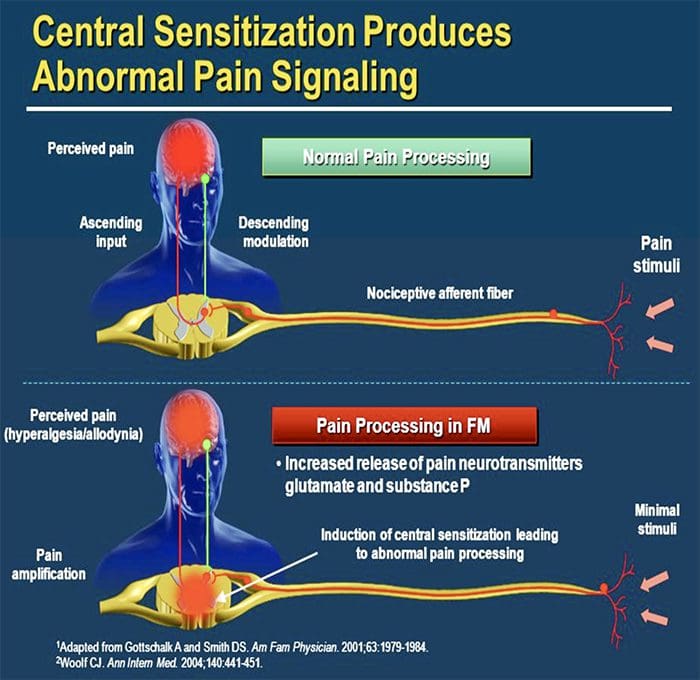
CENTRAL MECHANISMS
- As a consequence of ongoing spontaneous activity arising in the periphery, neurons develop an increased background activity, enlarged receptive fields and increased responses to afferent impulses, including normal tactile stimuli
This is known as…Central Sensitization!
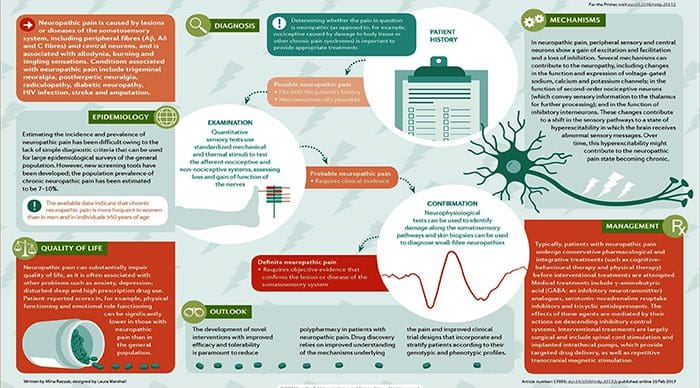 Chronic neuropathic pain is more frequent in women (8% versus 5.7% in men) and in patients >50 years of age (8.9% versus 5.6% in those <49 years of age), and most commonly affects the lower back and lower limbs, neck and upper limbs24. Lumbar and cervical painful radiculopathies are probably the most frequent cause of chronic neuropathic pain. Consistent with these data, a survey of >12,000 patients with chronic pain with both nociceptive and neuropathic pain types, referred to pain specialists in Germany, revealed that 40% of all patients experience at least some characteristics of neuropathic pain (such as burning sensations, numbness and tingling); patients with chronic back pain and radiculopathy were particularly affected25.
Chronic neuropathic pain is more frequent in women (8% versus 5.7% in men) and in patients >50 years of age (8.9% versus 5.6% in those <49 years of age), and most commonly affects the lower back and lower limbs, neck and upper limbs24. Lumbar and cervical painful radiculopathies are probably the most frequent cause of chronic neuropathic pain. Consistent with these data, a survey of >12,000 patients with chronic pain with both nociceptive and neuropathic pain types, referred to pain specialists in Germany, revealed that 40% of all patients experience at least some characteristics of neuropathic pain (such as burning sensations, numbness and tingling); patients with chronic back pain and radiculopathy were particularly affected25.

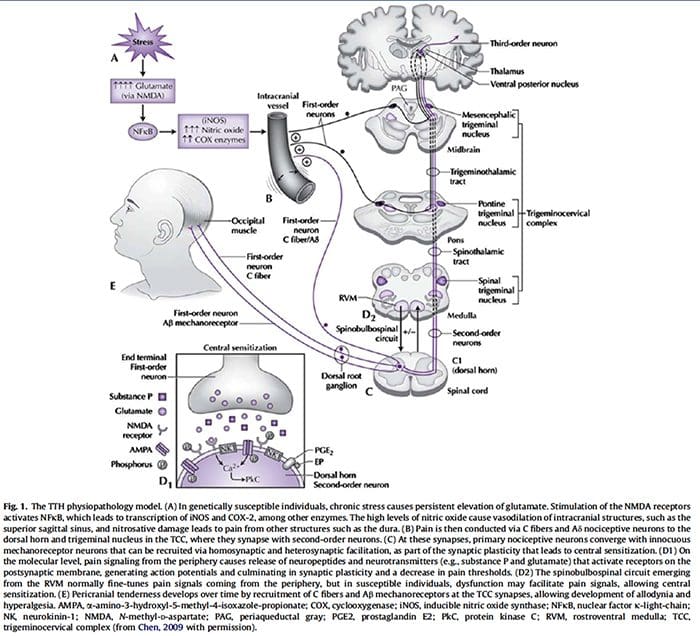
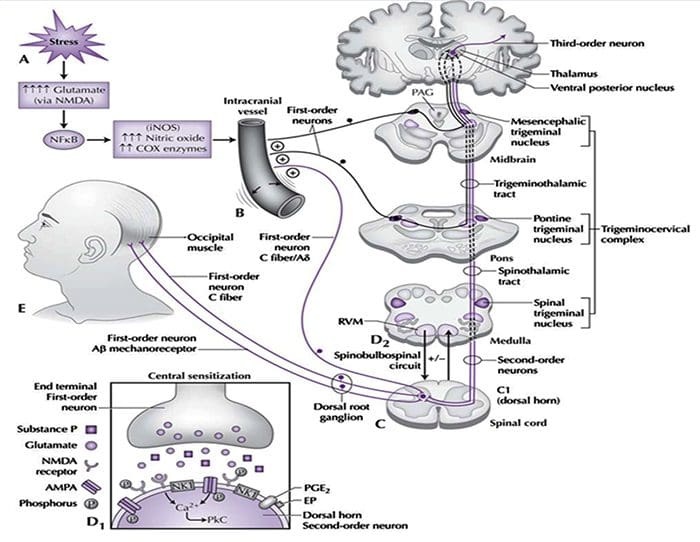
The contribution of clinical neurophysiology to the comprehension of the tension-type headache mechanisms.
Abstract
So far, clinical neurophysiological studies on tension-type headache (TTH) have been conducted with two main purposes: (1) to establish whether some neurophysiological parameters may act as markers of TTH, and (2) to investigate the physiopathology of TTH. With regard to the first point, the present results are disappointing, since some abnormalities found in TTH patients may be frequently observed also in migraineurs. On the other hand, clinical neurophysiology has played an important role in the debate about the pathogenesis of TTH. Studies on the exteroceptive suppression of the temporalis muscle contraction have detected a dysfunction of the brainstem excitability and of its suprasegmental control. A similar conclusion has been reached by using the trigeminocervical reflexes, whose abnormalities in TTH have suggested a reduced inhibitory activity of brainstem interneurons, reflecting abnormal endogenous pain control mechanisms. It is interesting that the neural excitability abnormality in TTH seems to be a generalized phenomenon, not limited to the cranial districts. Defective DNIC-like mechanisms have indeed been evidenced also in somatic districts by nociceptive flexion reflex studies. Unfortunately, most neurophysiological studies on TTH are marred by serious methodological flaws, which should be avoided in future researches, in order to better clarify the TTH mechanisms.


References:
 References:
References:Neurophysiology of arthritis pain. McDougall JJ1, Linton P.
https://www.researchgate.net/publication/232231610_Neurophysiology_of_Arthritis_Pain
Pain originating from the lumbar facet joints. van Kleef M1, Vanelderen P, Cohen SP, Lataster A, Van Zundert J, Mekhail N.
Neuropathic pain Luana Colloca,1 Taylor Ludman,1 Didier Bouhassira,2 Ralf Baron,3 Anthony H. Dickenson,4 David Yarnitsky,5Roy Freeman,6 Andrea Truini,7 Nadine Attal,8 Nanna B. Finnerup,9 Christopher Eccleston,10,11 Eija Kalso,12David L. Bennett,13 Robert H. Dworkin,14 and Srinivasa N. Raja15
The contribution of clinical neurophysiology to the comprehension of the tension-type headache mechanisms. Rossi P1, Vollono C, Valeriani M, Sandrini G.
Post Disclaimer
Professional Scope of Practice *
The information herein on "Facetogenic Pain, Headaches, Neuropathic Pain And Osteoarthritis" is not intended to replace a one-on-one relationship with a qualified health care professional or licensed physician and is not medical advice. We encourage you to make healthcare decisions based on your research and partnership with a qualified healthcare professional.
Blog Information & Scope Discussions
Welcome to El Paso's Premier Wellness, Personal Injury Care Clinic & Wellness Blog, where Dr. Alex Jimenez, DC, FNP-C, a Multi-State board-certified Family Practice Nurse Practitioner (FNP-BC) and Chiropractor (DC), presents insights on how our multidisciplinary team is dedicated to holistic healing and personalized care. Our practice aligns with evidence-based treatment protocols inspired by integrative medicine principles, similar to those on this site and our family practice-based chiromed.com site, and focuses on restoring health naturally for patients of all ages.
Our areas of multidisciplinary practice include Wellness & Nutrition, Chronic Pain, Personal Injury, Auto Accident Care, Work Injuries, Back Injury, Low Back Pain, Neck Pain, Migraine Headaches, Sports Injuries, Severe Sciatica, Scoliosis, Complex Herniated Discs, Fibromyalgia, Chronic Pain, Complex Injuries, Stress Management, Functional Medicine Treatments, and in-scope care protocols.
Our information scope is multidisciplinary, focusing on musculoskeletal and physical medicine, wellness, contributing etiological viscerosomatic disturbances within clinical presentations, associated somato-visceral reflex clinical dynamics, subluxation complexes, sensitive health issues, and functional medicine articles, topics, and discussions.
We provide and present clinical collaboration with specialists from various disciplines. Each specialist is governed by their professional scope of practice and their jurisdiction of licensure. We use functional health & wellness protocols to treat and support care for musculoskeletal injuries or disorders.
Our videos, posts, topics, and insights address clinical matters and issues that are directly or indirectly related to our clinical scope of practice.
Our office has made a reasonable effort to provide supportive citations and has identified relevant research studies that support our posts. We provide copies of supporting research studies upon request to regulatory boards and the public.
We understand that we cover matters that require an additional explanation of how they may assist in a particular care plan or treatment protocol; therefore, to discuss the subject matter above further, please feel free to ask Dr. Alex Jimenez, DC, APRN, FNP-BC, or contact us at 915-850-0900.
We are here to help you and your family.
Blessings
Dr. Alex Jimenez DC, MSACP, APRN, FNP-BC*, CCST, IFMCP, CFMP, ATN
email: [email protected]
Multidisciplinary Licensing & Board Certifications:
Licensed as a Doctor of Chiropractic (DC) in Texas & New Mexico*
Texas DC License #: TX5807, Verified: TX5807
New Mexico DC License #: NM-DC2182, Verified: NM-DC2182
Multi-State Advanced Practice Registered Nurse (APRN*) in Texas & Multi-States
Multi-state Compact APRN License by Endorsement (42 States)
Texas APRN License #: 1191402, Verified: 1191402 *
Florida APRN License #: 11043890, Verified: APRN11043890 *
Colorado License #: C-APN.0105610-C-NP, Verified: C-APN.0105610-C-NP
New York License #: N25929, Verified N25929
License Verification Link: Nursys License Verifier
* Prescriptive Authority Authorized
ANCC FNP-BC: Board Certified Nurse Practitioner*
Compact Status: Multi-State License: Authorized to Practice in 40 States*
Graduate with Honors: ICHS: MSN-FNP (Family Nurse Practitioner Program)
Degree Granted. Master's in Family Practice MSN Diploma (Cum Laude)
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)
(Licensed Medical Doctor)
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933
Licenses and Board Certifications:
MD: Medical Doctor
DC: Doctor of Chiropractic
APRNP: Advanced Practice Registered Nurse
FNP-BC: Family Practice Specialization (Multi-State Board Certified)
RN: Registered Nurse (Multi-State Compact License)
CFMP: Certified Functional Medicine Provider
MSN-FNP: Master of Science in Family Practice Medicine
MSACP: Master of Science in Advanced Clinical Practice
IFMCP: Institute of Functional Medicine
CCST: Certified Chiropractic Spinal Trauma
ATN: Advanced Translational Neutrogenomics
Memberships & Associations:
TCA: Texas Chiropractic Association: Member ID: 104311
AANP: American Association of Nurse Practitioners: Member ID: 2198960
ANA: American Nurse Association: Member ID: 06458222 (District TX01)
TNA: Texas Nurse Association: Member ID: 06458222
NPI: 1205907805
| Primary Taxonomy | Selected Taxonomy | State | License Number |
|---|---|---|---|
| No | 111N00000X - Chiropractor | NM | DC2182 |
| Yes | 111N00000X - Chiropractor | TX | DC5807 |
| Yes | 363LF0000X - Nurse Practitioner - Family | TX | 1191402 |
| Yes | 363LF0000X - Nurse Practitioner - Family | FL | 11043890 |
| Yes | 363LF0000X - Nurse Practitioner - Family | CO | C-APN.0105610-C-NP |
| Yes | 363LF0000X - Nurse Practitioner - Family | NY | N25929 |
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)*
(Licensed Medical Doctor)*
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933


 Again, We Welcome You.
Again, We Welcome You.
Comments are closed.