

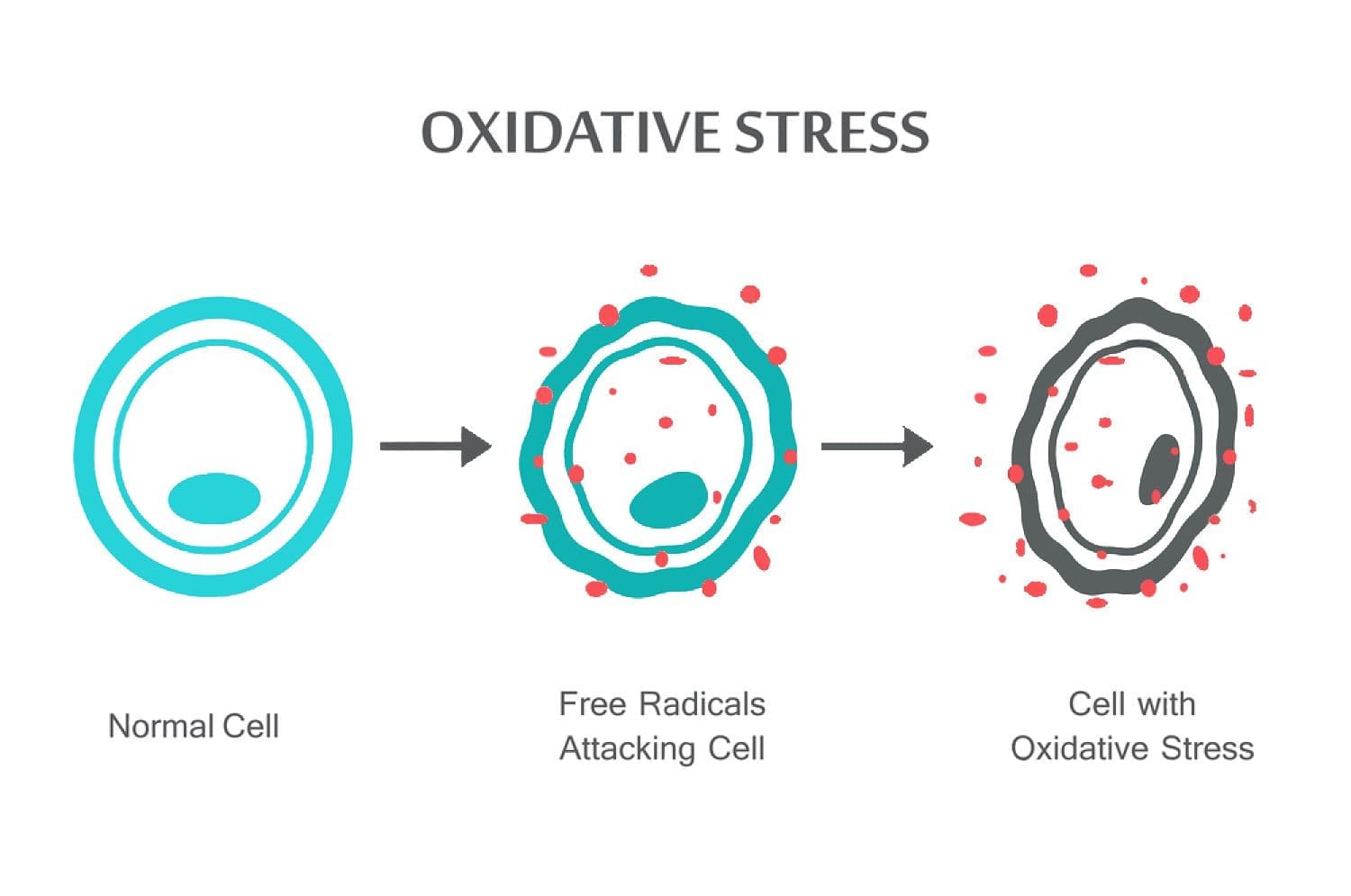
Science based Chiropractor Dr. Alexander Jimenez takes a look at oxidative stress, what it is, how it affects the body and the antioxidant defense to remedy the situation.
Esra Birben PhD,1 Umit Murat Sahiner MD,1 Cansin Sackesen MD,1 Serpil Erzurum MD,2 and Omer Kalayci, MD1
Abstract: Reactive oxygen species (ROS) are produced by living organisms as a result of normal cellular metabolism and environ- mental factors, such as air pollutants or cigarette smoke. ROS are highly reactive molecules and can damage cell structures such as carbohydrates, nucleic acids, lipids, and proteins and alter their functions. The shift in the balance between oxidants and antioxidants in favor of oxidants is termed “oxidative stress.†Regulation of reducing and oxidizing (redox) state is critical for cell viability, activation, proliferation, and organ function. Aerobic organisms have integrated antioxidant systems, which include enzymatic and non- enzymatic antioxidants that are usually effective in blocking harmful effects of ROS. However, in pathological conditions, the antioxidant systems can be overwhelmed. Oxidative stress contributes to many pathological conditions and diseases, including cancer, neurological disorders, atherosclerosis, hypertension, ischemia/perfusion, diabetes, acute respiratory distress syndrome, idiopathic pulmonary fibrosis, chronic obstructive pulmonary disease, and asthma. In this review, we summarize the cellular oxidant and antioxidant systems and discuss the cellular effects and mechanisms of the oxidative stress.
Key Words: antioxidant, oxidant, oxidative stress, reactive oxygen species, redox
(WAO Journal 2012; 5:9–19)
Reactive oxygen species (ROS) are produced by living organisms as a result of normal cellular metabolism. At low to moderate concentrations, they function in physiological cell processes, but at high concentrations, they produce adverse modifications to cell components, such as lipids, proteins, and DNA.1–6 The shift in balance between oxidant/ antioxidant in favor of oxidants is termed “oxidative stress.†Oxidative stress contributes to many pathological conditions, including cancer, neurological disorders,7–10 atherosclerosis, hypertension, ischemia/perfusion,11–14 diabetes, acute respiratory distress syndrome, idiopathic pulmonary fibrosis, chronic obstructive pulmonary disease,15 and asthma.16–21 Aerobic organisms have integrated antioxidant systems, which include enzymatic and nonenzymatic antioxidants that are usually effective in blocking harmful effects of ROS. However, in pathological conditions, the antioxidant systems can be overwhelmed. In this review, we summarize the cellular oxidant and antioxidant systems and regulation of the reducing and oxidizing (redox) state in health and disease states.
Table of Contents
OXIDANTS
Endogenous Sources of ROS
ROS are produced from molecular oxygen as a result of normal cellular metabolism. ROS can be divided into 2 groups: free radicals and nonradicals. Molecules containing one or more unpaired electrons and thus giving reactivity to the molecule are called free radicals. When 2 free radicals share their unpaired electrons, nonradical forms are created. The 3 major ROS that are of physiological significance are superoxide anion (O22.), hydroxyl radical ( OH), and hydro- gen peroxide (H2O2). ROS are summarized in Table 1.
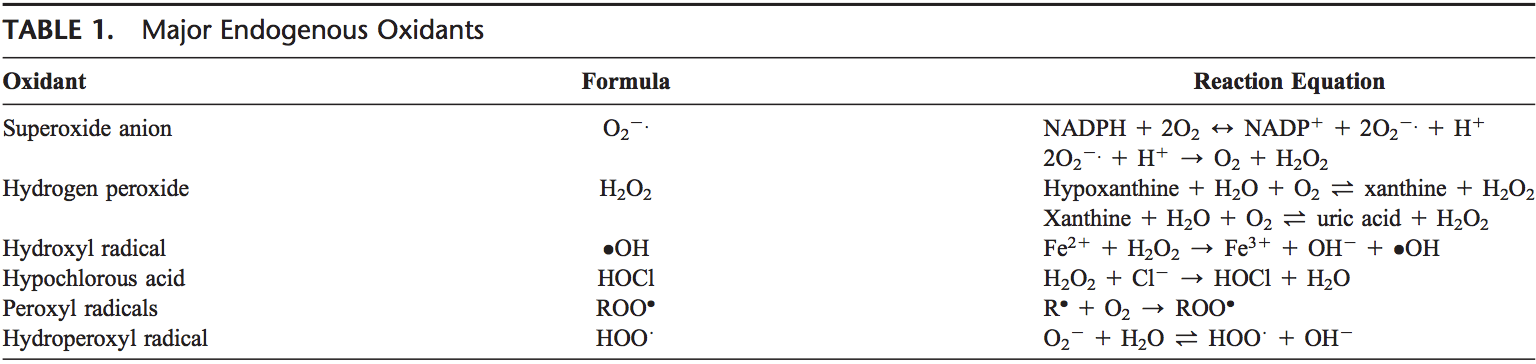 Superoxide anion is formed by the addition of 1 electron to the molecular oxygen.22 This process is mediated by nicotine adenine dinucleotide phosphate [NAD(P)H] oxidase or xanthine oxidase or by mitochondrial electron trans- port system. The major site for producing superoxide anion is the mitochondria, the machinery of the cell to produce adenosine triphosphate. Normally, electrons are transferred through mitochondrial electron transport chain for reduction of oxygen to water, but approximately 1 to 3% of all electrons leak from the system and produce superoxide. NAD(P)H oxidase is found in polymorphonuclear leukocytes, monocytes, and macrophages. Upon phagocytosis, these cells produce a burst of superoxide that lead to bactericidal activity. Superoxide is converted into hydrogen peroxide by the action of superoxide dismutases (SODs, EC 1.15.1.1). Hydrogen peroxide easily diffuses across the plasma membrane. Hydrogen peroxide is also produced by xanthine oxidase, amino acid oxidase, and NAD(P)H oxidase 23,24 and in peroxisomes by consumption of molecular oxygen in metabolic reactions. In a succession of reactions called Haber–Weiss and Fenton reactions,H2O2 can breakdown to OH2 in the presence of transmission metals like Fe21 or Cu21.25
Superoxide anion is formed by the addition of 1 electron to the molecular oxygen.22 This process is mediated by nicotine adenine dinucleotide phosphate [NAD(P)H] oxidase or xanthine oxidase or by mitochondrial electron trans- port system. The major site for producing superoxide anion is the mitochondria, the machinery of the cell to produce adenosine triphosphate. Normally, electrons are transferred through mitochondrial electron transport chain for reduction of oxygen to water, but approximately 1 to 3% of all electrons leak from the system and produce superoxide. NAD(P)H oxidase is found in polymorphonuclear leukocytes, monocytes, and macrophages. Upon phagocytosis, these cells produce a burst of superoxide that lead to bactericidal activity. Superoxide is converted into hydrogen peroxide by the action of superoxide dismutases (SODs, EC 1.15.1.1). Hydrogen peroxide easily diffuses across the plasma membrane. Hydrogen peroxide is also produced by xanthine oxidase, amino acid oxidase, and NAD(P)H oxidase 23,24 and in peroxisomes by consumption of molecular oxygen in metabolic reactions. In a succession of reactions called Haber–Weiss and Fenton reactions,H2O2 can breakdown to OH2 in the presence of transmission metals like Fe21 or Cu21.25
Fe31 + .O2 →Fe2 + O2 Haber Weiss
Fe2 + H2O2 →Fe3 + OH + .OH Fenton reaction
O 2 Â itself can also react with H2 O2 and generate OHÂ .26,27 Hydroxyl radical is the most reactive of ROS and can damage proteins, lipids, and carbohydrates and DNA. It can also start lipid peroxidation by taking an electron from polyunsaturated fatty acids.
Granulocytic enzymes further expand the reactivity of H2O2 via eosinophil peroxidase and myeloperoxidase (MPO). In activated neutrophils, H2O2 is consumed by MPO. In the presence of chloride ion, H2O2 is converted to hypochlorous acid (HOCl). HOCl is highly oxidative and plays an important role in killing of the pathogens in the airways.28 However, HOCl can also react with DNA and induce DNA–protein interactions and produce pyrimidine oxidation products and add chloride to DNA bases.29,30 Eosinophil peroxidase and MPO also contribute to the oxidative stress by modification of proteins by halogenations, nitration, and protein cross-links via tyrosyl radicals.31–33
Other oxygen-derived free radicals are the peroxyl radicals (ROO$ ). Simplest form of these radicals is hydro- peroxyl radical (HOO$ ) and has a role in fatty acid peroxidation. Free radicals can trigger lipid peroxidation chain reactions by abstracting a hydrogen atom from a side- chain methylene carbon. The lipid radical then reacts with oxygen to produce peroxyl radical. Peroxyl radical initiates a chain reaction and transforms polyunsaturated fatty acids into lipid hydroperoxides. Lipid hydroperoxides are very unstable and easily decompose to secondary products, such as aldehydes (such as 4-hydroxy-2,3-nonenal) and malondialdehydes (MDAs). Isoprostanes are another group of lipid peroxidation products that are generated via the peroxidation of arachidonic acid and have also been found to be elevated in plasma and breath condensates of asthmatics.34,35 Peroxidation of lipids disturbs the integrity of cell membranes and leads to rearrangement of membrane structure.
Hydrogen peroxide, superoxide radical, oxidized glutathione (GSSG), MDAs, isoprostanes, carbonyls, and nitrotyrosine can be easily measured from plasma, blood, or bronchoalveolar lavage samples as biomarkers of oxidation by standardized assays.
Exogenous Source of Oxidants
Cigarette Smoke
Cigarette smoke contains many oxidants and free radicals and organic compounds, such as superoxide and nitric oxide.36 In addition, inhalation of cigarette smoke into the lung also activates some endogenous mechanisms, such as accumulation of neutrophils and macrophages, which further increase the oxidant injury.
Ozone Exposure
Ozone exposure can cause lipid peroxidation and induce influx of neutrophils into the airway epithelium. Short-term exposure to ozone also causes the release of inflammatory mediators, such as MPO, eosinophil cationic proteins and also lactate dehydrogenase and albumin.37 Even in healthy subjects, ozone exposure causes a reduction in pulmonary functions.38 Cho et al39 have shown that particulate matter (mixture of solid particles and liquid droplets suspended in the air) catalyzes the reduction of oxygen.
Hyperoxia
Hyperoxia refers to conditions of higher oxygen levels than normal partial pressure of oxygen in the lungs or other body tissues. It leads to greater production of reactive oxygen and nitrogen species.40,41
Ionizing Radiation
Ionizing radiation, in the presence of O2, converts hydroxyl radical, superoxide, and organic radicals to hydrogen peroxide and organic hydroperoxides. These hydroperoxide species react with redox active metal ions, such as Fe and Cu, via Fenton reactions and thus induce oxidative stress.42,43 Narayanan et al44 showed that fibroblasts that were exposed to alpha particles had significant increases in intracellular O2 2. and H2O2 production via plasma membrane-bound NADPH oxidase.44 Signal transduction molecules, such as extracellular signal-regulated kinase 1 and 2 (ERK1/2), c-Jun N-terminal kinase (JNK), and p38, and transcription factors, such as activator protein-1 (AP-1), nuclear factor-kB (NF-kB), and p53, are activated, which result in the expression of radiation response–related genes.45–50 Ultraviolet A (UVA) photons trigger oxidative reactions by excitation of endogenous photosensitizers, such as porphyrins, NADPH oxidase, and riboflavins. 8-Oxo-7,8- dihydroguanine (8-oxoGua) is the main UVA-mediated DNA oxidation product formed by the oxidation of OH radical, 1-electron oxidants, and singlet oxygen that mainly reacts with guanine.51 The formation of guanine radical cation in isolated DNA has been shown to efficiently occur through the direct effect of ionizing radiation.52,53 After exposure to ionizing radiation, intracellular level of glutathione (GSH) decreases for a short term but then increases again.54
Heavy Metal Ions
Heavy metal ions, such as iron, copper, cadmium, mercury, nickel, lead, and arsenic, can induce generation of reactive radicals and cause cellular damage via depletion of enzyme activities through lipid peroxidation and reaction with nuclear proteins and DNA.55
One of the most important mechanisms of metal- mediated free radical generation is via a Fenton-type reaction. Superoxide ion and hydrogen peroxide can interact with transition metals, such as iron and copper, via the metal catalyzed Haber–Weiss/Fenton reaction to form OH radicals.
Metal31 1 $O2 /Metal21 1 O2 Haber Weiss Metal21 1 H2 O2 /Metal31 1 OH 2 1 $OH Fenton reaction
Besides the Fenton-type and Haber–Weiss-type mechanisms, certain metal ions can react directly with cellular molecules to generate free radicals, such as thiol radicals, or induce cell signaling pathways. These radicals may also react with other thiol molecules to generate O22.. O22. is converted to H2O2, which causes additional oxygen radical generation. Some metals, such as arsenite, induce ROS formation indirectly by activation of radical producing systems in cells.56
Arsenic is a highly toxic element that produces a variety of ROS, including superoxide (O2 2), singlet oxygen (1O2), peroxyl radical (ROO ), nitric oxide (NO ), hydrogen peroxide (H2O2), and dimethylarsinic peroxyl radicals [(CH3)2AsOO ].57–59 Arsenic (III) compounds can inhibit antioxidant enzymes, especially the GSH-dependent enzymes, such as glutathione-S-transferases (GSTs), glutathione peroxidase (GSH-Px), and GSH reductase, via bind- ing to their sulfhydryl (–SH) groups.60,61
Lead increases lipid peroxidation.62 Significant decreases in the activity of tissue SOD and brain GPx have been reported after lead exposure.63,64 Replacement of zinc, which serves as a cofactor for many enzymes by lead, leads to inactivation of such enzymes. Lead exposure may cause inhibition of GST by affecting tissue thiols.
ROS generated by metal-catalyzed reactions can mod- ify DNA bases. Three base substitutions, G / C, G / T, and C / T, can occur as a result of oxidative damage by metal ions, such as Fe21, Cu21, and Ni21. Reid et al65 showed that G / C was predominantly produced by Fe21 while C / T substitution was by Cu21 and Ni21.
ANTIOXIDANTS
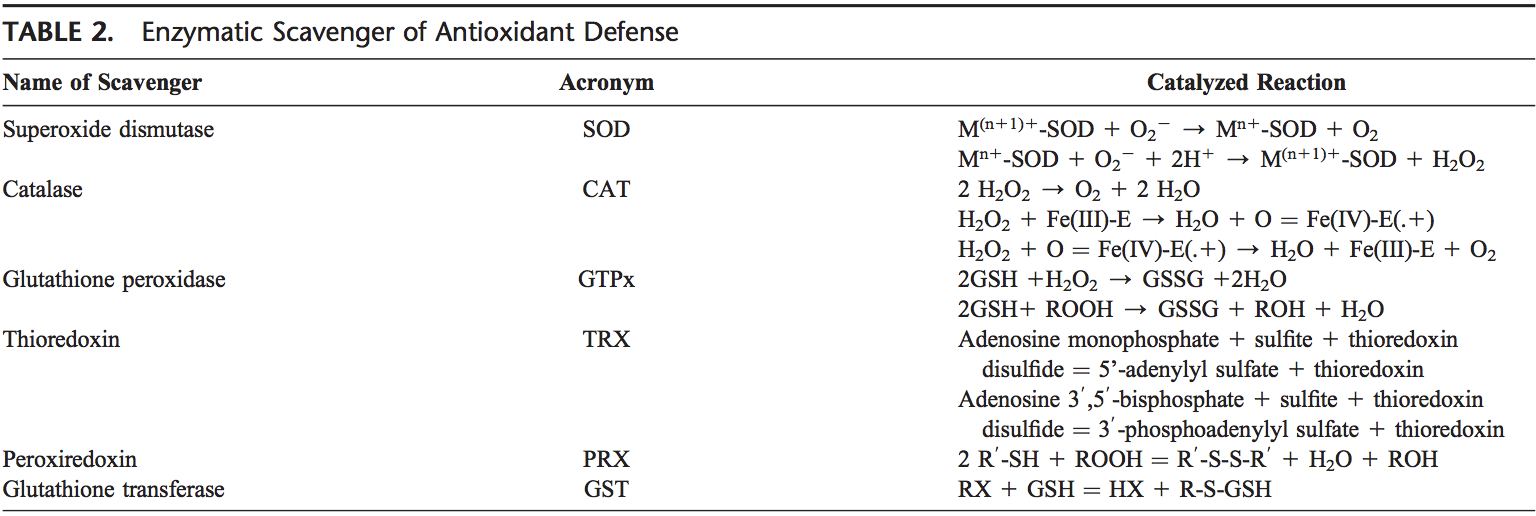
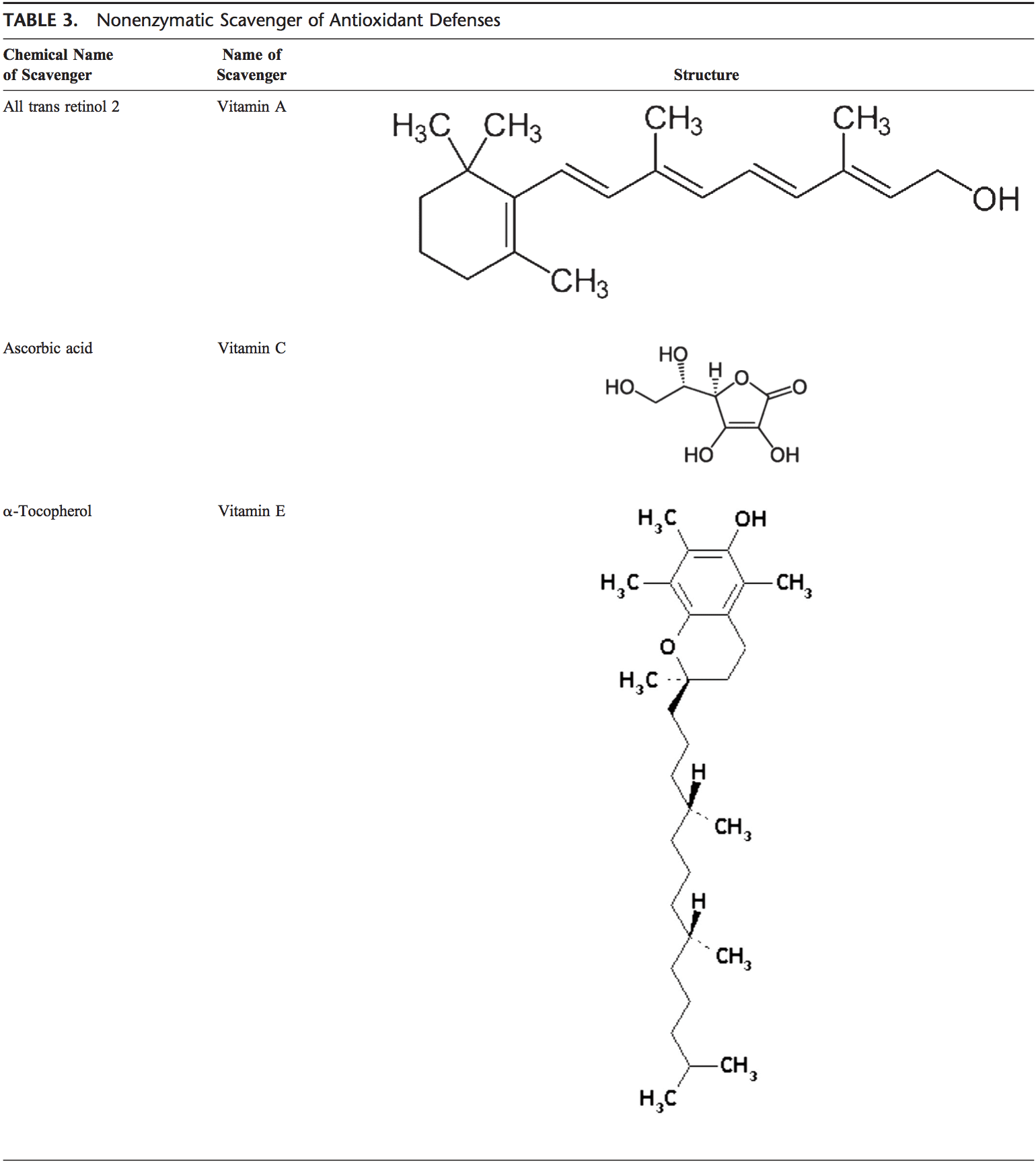
The human body is equipped with a variety of antioxidants that serve to counterbalance the effect of oxidants. For all practical purposes, these can be divided into 2 categories: enzymatic (Table 2) and nonenzymatic (Table 3).
 Enzymatic Antioxidants
Enzymatic Antioxidants
 Enzymatic Antioxidants
Enzymatic AntioxidantsThe major enzymatic antioxidants of the lungs are SODs (EC 1.15.1.11), catalase (EC 1.11.1.6), and GSH-Px (EC 1.11.1.9). In addition to these major enzymes, other antioxidants, including heme oxygenase-1 (EC 1.14.99.3), and redox proteins, such as thioredoxins (TRXs, EC 1.8.4.10), peroxiredoxins (PRXs, EC 1.11.1.15), and glutaredoxins, have also been found to play crucial roles in the pulmonary antioxidant defenses.
Since superoxide is the primary ROS produced from a variety of sources, its dismutation by SOD is of primary importance for each cell. All 3 forms of SOD, that is, CuZn- SOD, Mn-SOD, and EC-SOD, are widely expressed in the human lung. Mn-SOD is localized in the mitochondria matrix. EC-SOD is primarily localized in the extracellular matrix, especially in areas containing high amounts of type I collagen fibers and around pulmonary and systemic vessels. It has also been detected in the bronchial epithelium, alveolar epithelium, and alveolar macrophages.66,67 Overall, CuZn- SOD and Mn-SOD are generally thought to act as bulk scavengers of superoxide radicals. The relatively high EC-SOD level in the lung with its specific binding to the extracellular matrix components may represent a fundamental component of lung matrix protection.68
H2O2 that is produced by the action of SODs or the action of oxidases, such as xanthine oxidase, is reduced to water by catalase and the GSH-Px. Catalase exists as a tetra- mer composed of 4 identical monomers, each of which con- tains a heme group at the active site. Degradation of H2O2 is accomplished via the conversion between 2 conformations of catalase-ferricatalase (iron coordinated to water) and com- pound I (iron complexed with an oxygen atom). Catalase also binds NADPH as a reducing equivalent to prevent oxidative inactivation of the enzyme (formation of compound II) by H2O2 as it is reduced to water.69
Enzymes in the redox cycle responsible for the reduction of H2O2 and lipid hydroperoxides (generated as a result of membrane lipid peroxidation) include the GSH-Pxs.70 The GSH-Pxs are a family of tetrameric enzymes that contain the unique amino acid selenocysteine within the active sites and use low-molecular-weight thiols, such as GSH, to reduce H2O2 and lipid peroxides to their corresponding alcohols. Four GSH- Pxs have been described, encoded by different genes: GSH- Px-1 (cellular GSH-Px) is ubiquitous and reduces H2O2 and fatty acid peroxides, but not esterified peroxyl lipids.71 Esterified lipids are reduced by membrane-bound GSH-Px-4 (phospholipid hydroperoxide GSH-Px), which can use several different low-molecular-weight thiols as reducing equivalents. GSH-Px-2 (gastrointestinal GSH-Px) is localized in gastrointestinal epithelial cells where it serves to reduce dietary peroxides.72 GSH-Px-3 (extracellular GSH-Px) is the only member of the GSH-Px family that resides in the extracellular compartment and is believed to be one of the most important extracellular antioxidant enzyme in mammals. Of these, extracellular GSH-Px is most widely investigated in the human lung.73
In addition, disposal of H2O2 is closely associated with several thiol-containing enzymes, namely, TRXs (TRX1 and TRX2), thioredoxin reductases (EC 1.8.1.9) (TRRs), PRXs (which are thioredoxin peroxidases), and glutaredoxins.74
Two TRXs and TRRs have been characterized in human cells, existing in both cytosol and mitochondria. In the lung, TRX and TRR are expressed in bronchial and alveolar epithelium and macrophages. Six different PRXs have been found in human cells, differing in their ultrastructural compartmentalization. Experimental studies have revealed the importance of PRX VI in the protection of alveolar epithelium. Human lung expresses all PRXs in bronchial epithelium, alveolar epithelium, and macrophages.75 PRX V has recently been found to function as a peroxynitrite reductase,76 which means that it may function as a potential protective compound in the development of ROS-mediated lung injury.77
Common to these antioxidants is the requirement of NADPH as a reducing equivalent. NADPH maintains catalase in the active form and is used as a cofactor by TRX and GSH reductase (EC 1.6.4.2), which converts GSSG to GSH, a co-substrate for the GSH-Pxs. Intracellular NADPH, in turn, is generated by the reduction of NADP1 by glucose-6-phosphate dehydrogenase, the first and rate-limiting enzyme of the pen- tose phosphate pathway, during the conversion of glucose- 6-phosphate to 6-phosphogluconolactone. By generating NADPH, glucose-6-phosphate dehydrogenase is a critical determinant of cytosolic GSH buffering capacity (GSH/ GSSG) and, therefore, can be considered an essential, regulatory antioxidant enzyme.78,79
GSTs (EC 2.5.1.18), another antioxidant enzyme family, inactivate secondary metabolites, such as unsaturated aldehydes, epoxides, and hydroperoxides. Three major families of GSTs have been described: cytosolic GST, mitochondrial GST,80,81 and membrane-associated microsomal GST that has a role in eicosanoid and GSH metabolism.82 Seven classes of cytosolic GST are identified in mammalian, designated Alpha, Mu, Pi, Sigma, Theta, Omega, and Zeta.83–86 During non-stressed conditions, class Mu and Pi GSTs interact with kinases Ask1 and JNK, respectively, and inhibit these kinases.87–89 It has been shown that GSTP1 dissociates from JNK in response to oxidative stress.89 GSTP1 also physically interacts with PRX VI and leads to recovery of PRX enzyme activity via glutathionylation of the oxidized protein.90
Nonenzymatic Antioxidants
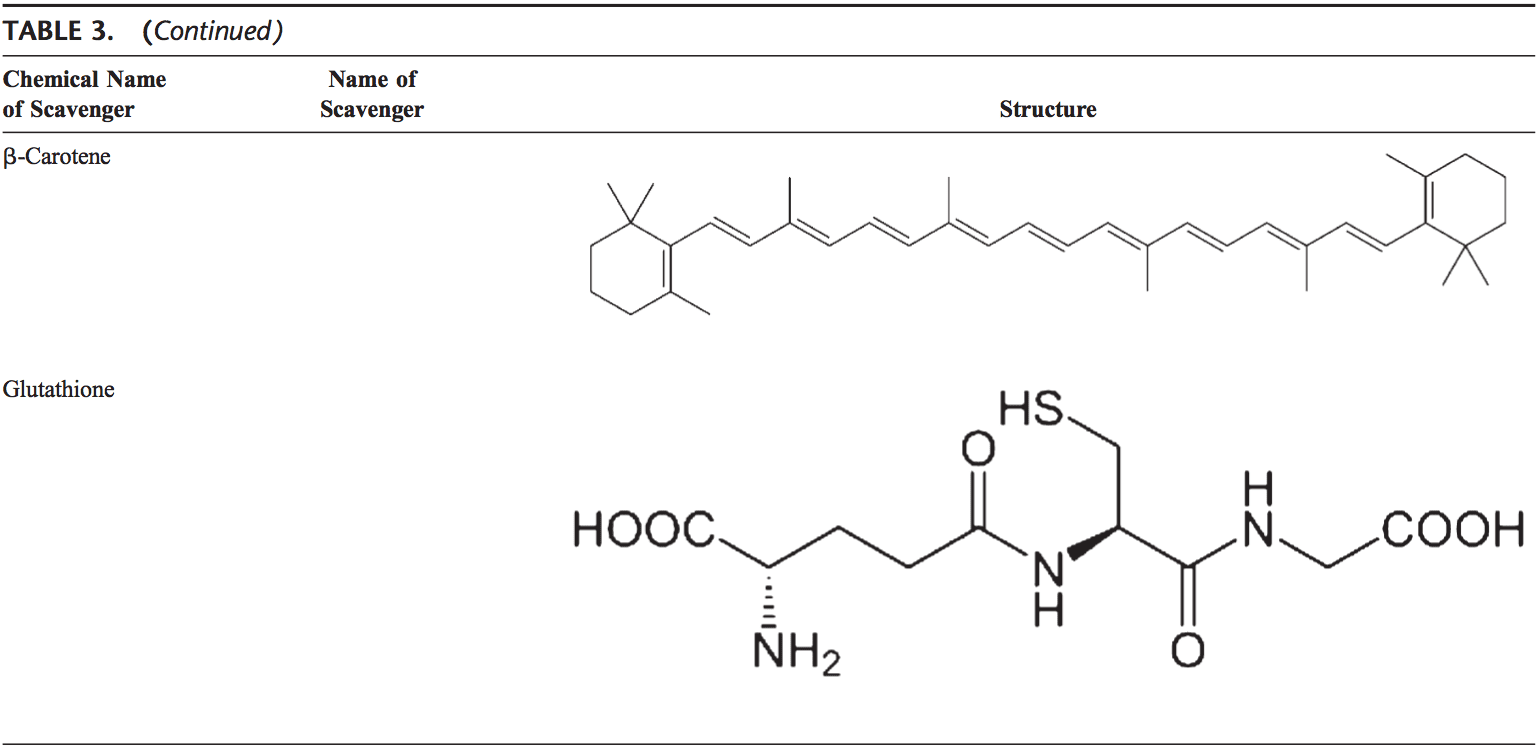
Nonenzymatic antioxidants include low-molecular-weight compounds, such as vitamins (vitamins C and E), b-carotene, uric acid, and GSH, a tripeptide (L-g-glutamyl-L-cysteinyl-L- glycine) that comprise a thiol (sulfhydryl) group.
Vitamin C (Ascorbic Acid)
Water-soluble vitamin C (ascorbic acid) provides intracellular and extracellular aqueous-phase antioxidant capacity primarily by scavenging oxygen free radicals. It converts vitamin E free radicals back to vitamin E. Its plasma levels have been shown to decrease with age.91,92
Vitamin E (a-Tocopherol)
Lipid-soluble vitamin E is concentrated in the hydrophobic interior site of cell membrane and is the principal defense against oxidant-induced membrane injury. Vitamin E donates electron to peroxyl radical, which is produced during lipid peroxidation. a-Tocopherol is the most active form of vitamin E and the major membrane-bound antioxidant in cell. Vitamin E triggers apoptosis of cancer cells and inhibits free radical formations.93
Glutathione
GSH is highly abundant in all cell compartments and is the major soluble antioxidant. GSH/GSSG ratio is a major determinant of oxidative stress. GSH shows its antioxidant effects in several ways.94 It detoxifies hydrogen peroxide and lipid peroxides via action of GSH-Px. GSH donates its electron to H2O2 to reduce it into H2O and O2. GSSG is again reduced into GSH by GSH reductase that uses NAD(P)H as the electron donor. GSH-Pxs are also important for the pro- tection of cell membrane from lipid peroxidation. Reduced glutathione donates protons to membrane lipids and protects them from oxidant attacks.95
GSH is a cofactor for several detoxifying enzymes, such as GSH-Px and transferase. It has a role in converting vitamin C and E back to their active forms. GSH protects cells against apoptosis by interacting with proapoptotic and antiapoptotic signaling pathways.94 It also regulates and activates several transcription factors, such as AP-1, NF-kB, and Sp-1.
Carotenoids (b-Carotene)
Carotenoids are pigments found in plants. Primarily, b-carotene has been found to react with peroxyl (ROO ), hydroxyl ( OH), and superoxide (O22.) radicals.96 Carotenoids show their antioxidant effects in low oxygen partial pressure but may have pro-oxidant effects at higher oxygen concentrations.97 Both carotenoids and retinoic acids (RAs) are capable of regulating transcription factors.98 b-Carotene inhibits the oxidant-induced NF-kB activation and interleukin (IL)-6 and tumor necrosis factor-a production. Carotenoids also affect apoptosis of cells. Antiproliferative effects of RA have been shown in several studies. This effect of RA is mediated mainly by retinoic acid receptors and vary among cell types. In mammary carcinoma cells, retinoic acid receptor was shown to trigger growth inhibition by inducing cell cycle arrest, apoptosis, or both.99,100
THE EFFECT OF OXIDATIVE STRESS: GENETIC, PHYSIOLOGICAL, & BIOCHEMICAL MECHANISMS
Oxidative stress occurs when the balance between antioxidants and ROS are disrupted because of either depletion of antioxidants or accumulation of ROS. When oxidative stress occurs, cells attempt to counteract the oxidant effects and restore the redox balance by activation or silencing of genes encoding defensive enzymes, tran- scription factors, and structural proteins.101,102 Ratio between oxidized and reduced glutathione (2GSH/GSSG) is one of the important determinants of oxidative stress in the body. Higher production of ROS in body may change DNA structure, result in modification of proteins and lipids, activation of several stress-induced transcription factors, and production of pro-inflammatory and anti-inflammatory cytokines.
Effects Of Oxidative Stress On DNA
ROS can lead to DNA modifications in several ways, which involves degradation of bases, single- or double- stranded DNA breaks, purine, pyrimidine or sugar-bound modifications, mutations, deletions or translocations, and cross-linking with proteins. Most of these DNA modifications (Fig. 1) are highly relevant to carcinogenesis, aging, and neurodegenerative, cardiovascular, and autoimmune diseases. Tobacco smoke, redox metals, and nonredox metals, such as iron, cadmium, chrome, and arsenic, are also involved in carcinogenesis and aging by generating free radicals or bind- ing with thiol groups. Formation of 8-OH-G is the best- known DNA damage occurring via oxidative stress and is a potential biomarker for carcinogenesis.
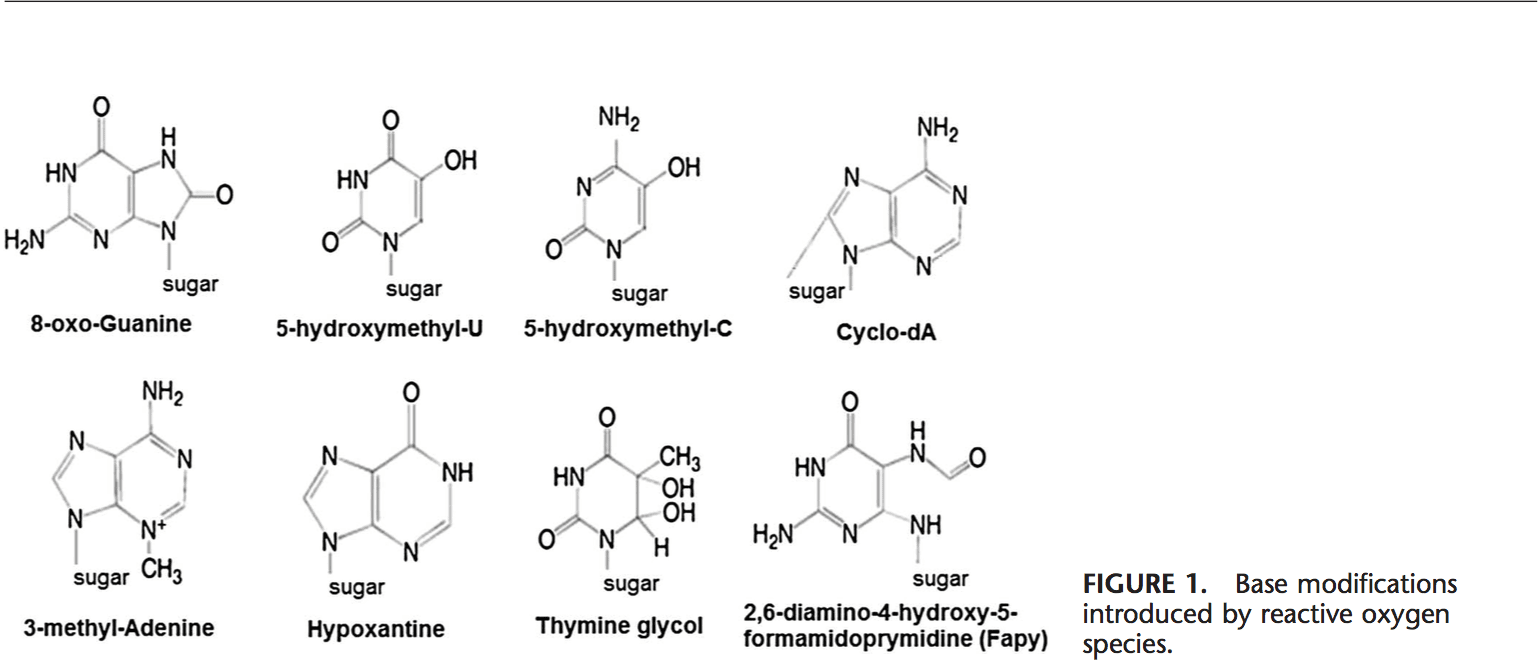 Promoter regions of genes contain consensus sequences for transcription factors. These transcription factor–binding sites contain GC-rich sequences that are susceptible for oxidant attacks. Formation of 8-OH-G DNA in transcription factor binding sites can modify binding of transcription factors and thus change the expression of related genes as has been shown for AP-1 and Sp-1 target sequences.103 Besides 8-OH-G, 8,59 -cyclo-29 -deoxyadenosine (cyclo-dA) has also been shown to inhibit transcription from a reporter gene in a cell system if located in a TATA box.104 The TATA-binding protein initiates transcription by changing the bending of DNA. The binding of TATA-binding protein may be impaired by the presence of cyclo-dA.
Promoter regions of genes contain consensus sequences for transcription factors. These transcription factor–binding sites contain GC-rich sequences that are susceptible for oxidant attacks. Formation of 8-OH-G DNA in transcription factor binding sites can modify binding of transcription factors and thus change the expression of related genes as has been shown for AP-1 and Sp-1 target sequences.103 Besides 8-OH-G, 8,59 -cyclo-29 -deoxyadenosine (cyclo-dA) has also been shown to inhibit transcription from a reporter gene in a cell system if located in a TATA box.104 The TATA-binding protein initiates transcription by changing the bending of DNA. The binding of TATA-binding protein may be impaired by the presence of cyclo-dA.
Oxidative stress causes instability of microsatellite (short tandem repeats) regions. Redox active metal ions, hydroxyl radicals increase microsatellite instability.105 Even though single-stranded DNA breaks caused by oxidant injury can easily be tolerated by cells, double-stranded DNA breaks induced by ionizing radiation can be a significant threat for the cell survival.106
Methylation at CpG islands in DNA is an important epigenetic mechanism that may result in gene silencing. Oxidation of 5-MeCyt to 5-hydroxymethyl uracil (5-OHMeUra) can occur via deamination/oxidation reactions of thymine or 5-hydroxymethyl cytosine intermediates.107 In addition to the modulating gene expression, DNA methylation also seems to affect chromatin organization.108 Aberrant DNA methylation patterns induced by oxidative attacks also affect DNA repair activity.
Effects Of Oxidative Stress On Lipids
ROS can induce lipid peroxidation and disrupt the membrane lipid bilayer arrangement that may inactivate membrane-bound receptors and enzymes and increase tissue permeability.109 Products of lipid peroxidation, such as MDA and unsaturated aldehydes, are capable of inactivating many cellular proteins by forming protein cross-linkages.110–112 4-Hydroxy-2-nonenal causes depletion of intracellular GSH and induces of peroxide production,113,114 activates epidermal growth factor receptor,115 and induces fibronectin production.116 Lipid peroxidation products, such as isoprostanes and thiobarbituric acid reactive substances, have been used as indirect biomarkers of oxidative stress, and increased levels were shown in the exhaled breath condensate or bronchoalveolar lavage fluid or lung of chronic obstructive pulmonary disease patients or smokers.117–119
Effects Of Oxidative Stress on Proteins
ROS can cause fragmentation of the peptide chain, alteration of electrical charge of proteins, cross-linking of proteins, and oxidation of specific amino acids and therefore lead to increased susceptibility to proteolysis by degradation by specific proteases.120 Cysteine and methionine residues in proteins are particularly more susceptible to oxidation.121 Oxidation of sulfhydryl groups or methionine residues of proteins cause conformational changes, protein unfolding, and degradation.8,121–123 Enzymes that have metals on or close to their active sites are especially more sensitive to metal catalyzed oxidation. Oxidative modification of enzymes has been shown to inhibit their activities.124,125
In some cases, specific oxidation of proteins may take place. For example, methionine can be oxidized methionine sulfoxide126 and phenylalanine to o-tyrosine127; sulfhydryl groups can be oxidized to form disulfide bonds;128 and carbonyl groups may be introduced into the side chains of proteins. Gamma rays, metal-catalyzed oxidation, HOCl, and ozone can cause formation of carbonyl groups.129
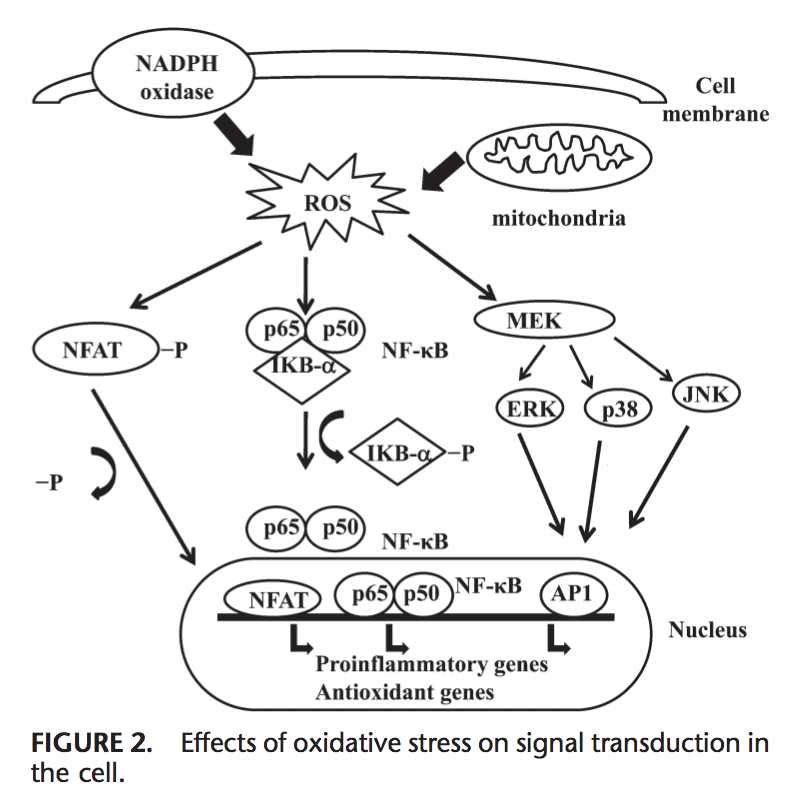
Effects of Oxidative Stress on Signal Transduction
ROS can induce expression of several genes involved in signal transduction.1,130 A high ratio for GSH/GSSG is important for the protection of the cell from oxidative dam- age. Disruption of this ratio causes activation of redox sensitive transcription factors, such as NF-kB, AP-1, nuclear factor of activated T cells and hypoxia-inducible factor 1 , that are involved in the inflammatory response. Activation of transcription factors via ROS is achieved by signal transduction cascades that transmit the information from outside to the inside of cell. Tyrosine kinase receptors, most of the growth factor receptors, such as epidermal growth factor receptor, vascular endothelial growth factor receptor, and receptor for platelet-derived growth factor, protein tyrosine phosphatases, and serine/threonine kinases are targets of ROS.131–133 Extra- cellular signal-regulated kinases, JNK, and p38, which are the members of mitogen-activated protein kinase family and involved in several processes in cell including proliferation, differentiation, and apoptosis, also can be regulated by oxidants.
Under oxidative stress conditions, cysteine residues in the DNA-binding site of c-Jun, some AP-1 subunits, and inhibitory k-B kinase undergo reversible S-glutathiolation. Glutaredoxin and TRX have been reported to play an important role in regulation of redox-sensitive signaling pathways, such as NF-kB and AP-1, p38 mitogen-activated protein kinase, and JNK.134–137
NF-kB can be activated in response to oxidative stress conditions, such as ROS, free radicals, and UV irradiation.138 Phosphorylation of IkB frees NF-kB and allows it to enter the nucleus to activate gene transcription.139 A number of kinases have been reported to phosphorylate IkBs at the serine residues. These kinases are the targets of oxidative signals for activation of NF-kB.140 Reducing agents enhance NF-kB DNA binding, whereas oxidizing agents inhibit DNA binding of NF-kB. TRX may exert 2 opposite actions in regulation of NF-kB: in the cytoplasm, it blocks degradation of IkB and inhibits NF-kB activation but enhances NF-kB DNA binding in the nucleus.141 Activation of NF-kB via oxidation-related degradation of IkB results in the activation of several antioxidant defense–related genes. NF-kB regulates the expression of several genes that participate in immune response, such as IL-1b, IL-6, tumor necrosis factor-a, IL-8, and several adhesion molecules.142,143 NF-kB also regulates angiogenesis and proliferation and differentiation of cells.
AP-1 is also regulated by redox state. In the presence of H2O2, some metal ions can induce activation of AP-1. Increase in the ratio of GSH/GSSG enhances AP-1 binding while GSSG inhibits the DNA binding of AP-1.144 DNA binding of the Fos/Jun heterodimer is increased by the reduction of a single conserved cysteine in the DNA-binding domain of each of the proteins,145 while DNA binding of AP-1 can be inhibited by GSSG in many cell types, suggesting that disulphide bond formation by cysteine residues inhibits AP-1 DNA binding.146,147 Signal transduction via oxidative stress is summarized in Figure 2.
CONCLUSIONS
Oxidative stress can arise from overproduction of ROS by metabolic reactions that use oxygen and shift the balance between oxidant/antioxidant statuses in favor of the oxidants. ROS are produced by cellular metabolic activities and environmental factors, such as air pollutants or cigarette smoke. ROS are highly reactive molecules because of unpaired electrons in their structure and react with several biological macromolecules in cell, such as carbohydrates, nucleic acids, lipids, and proteins, and alter their functions. ROS also affects the expression of several genes by upregulation of redox-sensitive transcription factors and chromatin remodeling via alteration in histone acetylation/ deacetylation. Regulation of redox state is critical for cell viability, activation, proliferation, and organ function.
References:
REFERENCES
- Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40.
- Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 3rd ed. New York: Oxford University Press;1999.
- Marnett LJ. Lipid peroxidationdDNA damage by malondialdehyde. Mutat Res. 1999;424:83–95.
- Siems WG, Grune T, Esterbauer H. 4-Hydroxynonenal formation during ischemia and reperfusion of rat small intestine.  Life Sci. 1995;57:785–789.
- Stadtman ER. Role of oxidant species in aging. Curr Med Chem. 2004;11:1105–1112.
- Wang MY, Dhingra K, Hittelman WN, Liehr JG, deAndrade M, Li DH. Lipid peroxidation-induced putative malondialdehyde–DNA adducts in human breast tissues. Cancer Epidemiol Biomarkers Prev. 1996;5:705–710.
- Jenner P. Oxidative stress in Parkinson’s disease. Ann Neurol. 2003;53: S26–S36.
- Lyras L, Cairns NJ, Jenner A, Jenner P, Halliwell B. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer’s disease. J Neurochem. 1997;68:2061–2069.
- Sayre LM, Smith MA, Perry G. Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Curr Med Chem. 2001;8:721–738.
- Toshniwal PK, Zarling EJ. Evidence for increased lipid peroxidation in multiple sclerosis. Neurochem Res. 1992;17:205–207.
- Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens. 2000;18:655–673.
- Kasparova S, Brezova V, Valko M, Horecky J, Mlynarik V, et al. Study of the oxidative stress in a rat model of chronic brain hypoperfusion. Neurochem Int. 2005;46:601–611.
- Kerr S, Brosnan MJ, McIntyre M, Reid JL, Dominiczak AF, Hamilton CA. Superoxide anion production is increased in a model of genetic hypertension: role of the endothelium. Hypertension. 1999;33:1353–1358.
- Kukreja RC, Hess ML. The oxygen free-radical system: from equations through membrane–protein interactions to cardiovascular injury and protection. Cardiovasc Res. 1992;26:641–655.
- Asami S, Manabe H, Miyake J, Tsurudome Y, Hirano T, et al. Cigarette smoking induces an increase in oxidative DNA damage, 8-hydroxydeoxyguanosine, in a central site of the human lung. Carcinogenesis. 1997;18:1763–1766.
- Andreadis AA, Hazen SL, Comhair SA, Erzurum SC. Oxidative and nitrosative events in asthma. Free Radic Biol Med. 2003;35:213–225.
- Comhair SA, Ricci KS, Arroliga M, Lara AR, Dweik RA, et al. Correlation of systemic superoxide dismutase deficiency to airflow obstruction in asthma. Am J Respir Crit Care Med. 2005;172:306–313.
- Comhair SA, Xu W, Ghosh S, Thunnissen FB, Almasan A, et al. Superoxide dismutase inactivation in pathophysiology of asthmatic airway remodeling and reactivity. Am J Pathol. 2005;166:663–674.
-
Dut R, Dizdar EA, Birben E, Sackesen C, Soyer OU, Besler T, Kalayci O. Oxidative stress and its determinants in the airways of children with asthma. Allergy. 2008;63:1605–1609.
-
Ercan H, Birben E, Dizdar EA, Keskin O, Karaaslan C, et al. Oxidative stress and genetic and epidemiologic determinants of oxidant injury in childhood asthma. J Allergy Clin Immunol. 2006;118:1097–1104.
- Fitzpatrick AM, Teague WG, Holguin F, Yeh M, Brown LA. Severe Asthma Research Program. Airway glutathione homeostasis is altered in children with severe asthma: evidence for oxidant stress. J Allergy Clin Immunol. 2009;123:146–152.
- Miller DM, Buettner GR, Aust SD. Transition metals as catalysts of “autoxidation” reactions. Free Radic Biol Med. 1990;8:95–108.
- Dupuy C, Virion A, Ohayon R, Kaniewski J, Dème D, Pommier J. Mechanism of hydrogen peroxide formation catalyzed by NADPH oxidase in thyroid plasma membrane. J Biol Chem. 1991;266:3739–3743.
- Granger DN. Role of xanthine oxidase and granulocytes in ischemiareperfusion injury. Am J Physiol. 1988;255:H1269–H1275.
- Fenton HJH. Oxidation of tartaric acid in the presence of iron. J Chem Soc. 1984;65:899–910.
- Haber F, Weiss JJ. The catalytic decomposition of hydrogen peroxide by iron salts. Proc R Soc Lond Ser A. 1934;147:332–351.
- Liochev SI, Fridovich I. The Haber–Weiss cycled70 years later: an alternative view. Redox Rep. 2002;7:55–57.
- Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598–625.
- Whiteman M, Jenner A, Halliwell B. Hypochlorous acid-induced base modifications in isolated calf thymus DNA. Chem Res Toxicol. 1997;10:1240–1246.
- Kulcharyk PA, Heinecke JW. Hypochlorous acid produced by the myeloperoxidase system of human phagocytes induces covalent cross-links between DNA and protein. Biochemistry. 2001;40:3648–3656.
- Brennan ML, Wu W, Fu X, Shen Z, Song W, et al. A tale of two controversies: defining both the role of peroxidases in nitrotyrosine formation in vivo using eosinophil peroxidase and myeloperoxidasedeficient mice, and the nature of peroxidase-generated reactive nitrogen species. J Biol Chem. 2002;277:17415–17427.
- Denzler KL, Borchers MT, Crosby JR, Cieslewicz G, Hines EM, et al. Extensive eosinophil degranulation and peroxidase-mediated oxidation of airway proteins do not occur in a mouse ovalbumin-challenge model of pulmonary inflammation. J Immunol. 2001;167:1672–1682.
- van Dalen CJ, Winterbourn CC, Senthilmohan R, Kettle AJ. Nitrite as a substrate and inhibitor of myeloperoxidase. Implications for nitration and hypochlorous acid production at sites of inflammation. J Biol Chem. 2000;275:11638–11644.
- Wood LG, Fitzgerald DA, Gibson PG, Cooper DM, Garg ML. Lipid peroxidation as determined by plasma isoprostanes is related to disease severity in mild asthma. Lipids. 2000;35:967–974.
- Montuschi P, Corradi M, Ciabattoni G, Nightingale J, Kharitonov SA, Barnes PJ. Increased 8-isoprostane, a marker of oxidative stress, in exhaled condensate of asthma patients. Am J Respir Crit Care Med. 1999;160:216–220.
- Church DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect. 1985;64:111–126.
- Hiltermann JT, Lapperre TS, van Bree L, Steerenberg PA, Brahim JJ, et al. Ozone-induced inflammation assessed in sputum and bronchial lavage fluid from asthmatics: a new noninvasive tool in epidemiologic studies on air pollution and asthma. Free Radic Biol Med. 1999;27:1448–1454.
- Nightingale JA, Rogers DF, Barnes PJ. Effect of inhaled ozone on exhaled nitric oxide, pulmonary function, and induced sputum in normal and asthmatic subjects. Thorax. 1999;54:1061–1069.
- Cho AK, Sioutas C, Miguel AH, Kumagai Y, Schmitz DA, et al. Redox activity of airborne particulate matter at different sites in the Los Angeles Basin. Environ Res. 2005;99:40–47.
- Comhair SA, Thomassen MJ, Erzurum SC. Differential induction of extracellular glutathione peroxidase and nitric oxide synthase 2 in airways of healthy individuals exposed to 100% O(2) or cigarette smoke. Am J Respir Cell Mol Biol. 2000;23:350–354.
- Matthay MA, Geiser T, Matalon S, Ischiropoulos H. Oxidant-mediated lung injury in the acute respiratory distress syndrome. Crit Care Med. 1999;27:2028–2030.
- Biaglow JE, Mitchell JB, Held K. The importance of peroxide and superoxide in the X-ray response. Int J Radiat Oncol Biol Phys. 1992;22:665–669.
- Chiu SM, Xue LY, Friedman LR, Oleinick NL. Copper ion-mediated sensitization of nuclear matrix attachment sites to ionizing radiation. Biochemistry. 1993;32:6214–6219.
- Narayanan PK, Goodwin EH, Lehnert BE. Alpha particles initiate biological production of superoxide anions and hydrogen peroxide in human cells. Cancer Res. 1997;57:3963–3971.
- Tuttle SW, Varnes ME, Mitchell JB, Biaglow JE. Sensitivity to chemical oxidants and radiation in CHO cell lines deficient in oxidative pentose cycle activity. Int J Radiat Oncol Biol Phys. 1992;22: 671–675.
- Guo G, Yan-Sanders Y, Lyn-Cook BD, Wang T, Tamae D, et al. Manganese
superoxide dismutase-mediated gene expression in radiationinduced
adaptive responses. Mol Cell Biol. 2003;23:2362–2378. - Azzam EI, de Toledo SM, Spitz DR, Little JB. Oxidative metabolism
modulates signal transduction and micronucleus formation in bystander
cells from a-particle irradiated normal human fibroblasts. Cancer Res.
2002;62:5436–5442. - Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB.
Ionizing radiation-induced, mitochondria-dependent generation of reactive
oxygen/nitrogen. Cancer Res. 2001;61:3894–3901. - Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK pathways in
radiation responses. Oncogene. 2003;22:5885–5896. - Wei SJ, Botero A, Hirota K, Bradbury CM, Markovina S, et al. Thioredoxin
nuclear translocation and interaction with redox factor-1 activates the AP-1 transcription factor in response to ionizing radiation. Cancer Res. 2000;60:6688–6695. - Cadet J, Douki T, Gasparutto D, Ravanat JL. Oxidative damage to DNA: formation, measurement and biochemical features. Mutat Res. 2003;531:5–23.
- Yokoya A, Cunniffe SM, O’Neill P. Effect of hydration on the induction of strand breaks and base lesions in plasmid DNA films by gammaradiation. J Am Chem Soc. 2002;124:8859–8866.
- Janssen YM, Van Houten B, Borm PJ, Mossman BT. Cell and tissue responses to oxidative damage. Lab Invest. 1993;69:261–274.
- Iwanaga M, Mori K, Iida T, Urata Y, Matsuo T, et al. Nuclear factor kappa B dependent induction of gamma glutamylcysteine synthetase by ionizing radiation in T98G human glioblastoma cells. Free Radic Biol Med. 1998;24:1256–1268.
- Stohs SJ, Bagchi D. Oxidative mechanisms in the toxicity of metal ions. Free Radic Biol Med. 1995;18:321–336.
- Leonard SS, Harris GK, Shi X. Metal-induced oxidative stress and signal transduction. Free Radic Biol Med. 2004;37:1921–1942.
- Shi H, Shi X, Liu KJ. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol Cell Biochem. 2004;255:67–78.
- Pi J, Horiguchi S, Sun Y, Nikaido M, Shimojo N, Hayashi T. A potential mechanism for the impairment of nitric oxide formation caused by prolonged oral exposure to arsenate in rabbits. Free Radic Biol Med.2003;35:102–113.
- Rin K, Kawaguchi K, Yamanaka K, Tezuka M, Oku N, Okada S. DNAstrand breaks induced by dimethylarsinic acid, a metabolite of inorganic arsenics, are strongly enhanced by superoxide anion radicals. Biol Pharm Bull. 1995;18:45–58.
- Waalkes MP, Liu J, Ward JM, Diwan LA. Mechanisms underlying arsenic carcinogenesis: hypersensitivity of mice exposed to inorganic arsenic during gestation. Toxicology. 2004;198:31–38.
- Schiller CM, Fowler BA, Woods JS. Effects of arsenic on pyruvate dehydrogenase activation. Environ Health Perspect. 1977;19:205–207.
- Monterio HP, Bechara EJH, Abdalla DSP. Free radicals involvement in neurological porphyrias and lead poisoning. Mol Cell Biochem. 1991;103:73–83.
- Tripathi RM, Raghunath R, Mahapatra S. Blood lead and its effect on Cd, Cu, Zn, Fe and hemoglobin levels of children. Sci Total Environ. 2001;277:161–168.
- Nehru B, Dua R. The effect of dietary selenium on lead neurotoxicity. J Environ Pathol Toxicol Oncol. 1997;16:47–50.
- Reid TM, Feig DI, Loeb LA. Mutagenesis by metal-induced oxygen radicals. Environ Health Perspect. 1994;102(suppl 3):57–61.
- Kinnula VL, Crapo JD. Superoxide dismutases in the lung and human lung diseases. Am J Respir Crit Care Med. 2003;167:1600–1619.
-
Kinnula VL. Production and degradation of oxygen metabolites during inflammatory states in the human lung. Curr Drug Targets Inflamm Allergy. 2005;4:465–470.
-
Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002;33:337–349.
- Kirkman HN, Rolfo M, Ferraris AM, Gaetani GF. Mechanisms of protection of catalase by NADPH. Kinetics and stoichiometry. J Biol Chem. 1999;274:13908–13914.
- Flohé L. Glutathione peroxidase. Basic Life Sci. 1988;49:663–668.
- Arthur JR. The glutathione peroxidases. Cell Mol Life Sci. 2000;57:1825–1835.
- Chu FF, Doroshow JH, Esworthy RS. Expression, characterization, and tissue distribution of a new cellular selenium-dependent glutathione peroxidase, GSHPx-GI. J Biol Chem. 1993;268:2571–2576.
- Comhair SA, Bhathena PR, Farver C, Thunnissen FB, Erzurum SC. Extracellular glutathione peroxidase induction in asthmatic lungs: evidence for redox regulation of expression in human airway epithelial cells. FASEB J. 2001;15:70–78.
- Gromer S, Urig S, Becker K. The thioredoxin systemdfrom science to clinic. Med Res Rev. 2004;24:40–89.
- Kinnula VL, Lehtonen S, Kaarteenaho-Wiik R, Lakari E, Pääkkö P, et al. Cell specific expression of peroxiredoxins in human lung and pulmonary sarcoidosis. Thorax. 2002;57:157–164.
- Dubuisson M, Vander Stricht D, Clippe A, Etienne F, Nauser T, et al. Human peroxiredoxin 5 is a peroxynitrite reductase. FEBS Lett. 2004;571:161–165.
- Holmgren A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid Redox Signal. 2000;2:811–820.
- Dickinson DA, Forman HJ. Glutathione in defense and signaling: lessons from a small thiol. Ann N Y Acad Sci. 2002;973:488–504.
- Sies H. Glutathione and its role in cellular functions. Free Radic Biol Med. 1999;27:916–921.
- Ladner JE, Parsons JF, Rife CL, Gilliland GL, Armstrong RN. Parallel evolutionary pathways for glutathione transferases: structure and mechanism of the mitochondrial class kappa enzyme rGSTK1-1. Biochemistry. 2004;43:52–61.
- Robinson A, Huttley GA, Booth HS, Board PG. Modelling and bioinformatics studies of the human kappa class glutathione transferase predict a novel third transferase family with homology to prokaryotic 2-hydroxychromene-2-carboxylate isomerases. Biochem J. 2004;379:541–552.
- Jakobsson P-J, Morgenstern R, Mancini J, Ford-Hutchinson A, Persson B. Common structural features of MAPEGda widespread superfamily of membrane associated proteins with highly divergent functions in eicosanoid and glutathione metabolism. Protein Sci. 1999;8:689–692.
- Hayes JD, Pulford DJ. The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol. 1995;30:445–600.
- Armstrong RN. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem Res Toxicol. 1997;10:2–18.
- Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic Res. 1999;31:273–300.
- Sheehan D, Meade G, Foley VM, Dowd CA. Structure, function and evolution of glutathione transferases: implications for classification of nonmammalian members of an ancient enzyme superfamily. Biochem J. 2001;360:1–16.
- Cho S-G, Lee YH, Park H-S, Ryoo K, Kang KW, et al. Glutathione S-transferase Mu modulates the stress activated signals by suppressing apoptosis signal-regulating kinase 1. J Biol Chem. 2001;276:12749–12755.
- Dorion S, Lambert H, Landry J. Activation of the p38 signaling pathway by heat shock involves the dissociation of glutathione S-transferase Mu from Ask1. J Biol Chem. 2002;277:30792–30797.
- Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, et al. Regulation of JNK signalling by GSTp. EMBO J. 1999;18:1321–1334.
- Manevich Y, Feinstein SI, Fisher AB. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pGST. Proc Natl Acad Sci U S A. 2004;101:3780–3785.
- Bunker VW. Free radicals, antioxidants and ageing. Med Lab Sci. 1992;49:299–312.
- Mezzetti A, Lapenna D, Romano F, Costantini F, Pierdomenico SD, et al. Systemic oxidative stress and its relationship with age and illness. J Am Geriatr Soc. 1996;44:823–827.
- White E, Shannon JS, Patterson RE. Relationship between vitamin and
calcium supplement use and colon cancer. Cancer Epidemiol Biomarkers Prev. 1997;6:769–774. - Masella R, Di Benedetto R, Vari R, Filesi C, Giovannini C. Novel mechanisms of natural antioxidant compounds in biological systems: involvement of glutathione and glutathione-related enzymes. J Nutr Biochem. 2005;16:577–586.
- Curello S, Ceconi C, Bigoli C, Ferrari R, Albertini A, Guarnieri C. Changes in the cardiac glutathione status after ischemia and reperfusion. Experientia. 1985;41:42–43.
- El-Agamey A, Lowe GM, McGarvey DJ, Mortensen A, Phillip DM, Truscott TG. Carotenoid radical chemistry and antioxidant/pro-oxidant properties. Arch Biochem Biophys. 2004;430:37–48.
- Rice-Evans CA, Sampson J, Bramley PM, Holloway DE. Why do we expect carotenoids to be antioxidants in vivo? Free Radic Res. 1997;26:381–398.
- Niles RM. Signaling pathways in retinoid chemoprevention and treatment of cancer. Mutat Res. 2004;555:81–96.
- Donato LJ, Noy N. Suppression of mammary carcinoma growth by retinoic acid: proapoptotic genes are targets for retinoic acid receptor and cellular retinoic acid-binding protein II signaling. Cancer Res. 2005;65:8193–8199.
- Niizuma H, Nakamura Y, Ozaki T, Nakanishi H, Ohira M, et al. Bcl-2 is a key regulator for the retinoic acid-induced apoptotic cell death in neuroblastoma. Oncogene. 2006;25:5046–5055.
- Dalton TP, Shertzer HG, Puga A. Regulation of gene expression by reactive oxygen. Ann Rev Pharmacol Toxicol. 1999;39:67–101.
- Scandalios JG. Genomic responses to oxidative stress. In: Meyers RA, ed. Encyclopedia of Molecular Cell Biology and Molecular Medicine. Vol 5. 2nd ed. Weinheim, Germany: Wiley-VCH; 2004: 489–512.
- Ghosh R, Mitchell DL. Effect of oxidative DNA damage in promoter elements on transcription factor binding. Nucleic Acids Res. 1999;27:3213–3218.
- Marietta C, Gulam H, Brooks PJ. A single 8, 50-cyclo-20-deoxyadenosine lesion in a TATA box prevents binding of the TATA binding protein and strongly reduces transcription in vivo. DNA Repair (Amst). 2002;1:967–975.
- Jackson AL, Chen R, Loeb LA. Induction of microsatellite instability
by oxidative DNA damage. Proc Natl Acad Sci U S A. 1998;95:12468–12473. - Caldecott KW. Protein-protein interactions during mammalian DNA single-strand break repair. Biochem Soc Trans. 2003;31:247–251.
- Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17:1195–1214.
- Jones PL, Wolffe AP. Relationships between chromatin organization and DNA methylation in determining gene expression. Semin Cancer Biol. 1999;9:339–347.
- Girotti AW. Mechanisms of lipid peroxidation. J Free Radic Biol Med. 1985;1:87–95.
- Siu GM, Draper HH. Metabolism of malonaldehyde in vivo and in vitro. Lipids. 1982;17:349–355.
- Esterbauer H, Koller E, Slee RG, Koster JF. Possible involvement of the lipid-peroxidation product 4-hydroxynonenal in the formation of fluorescent chromolipids. Biochem J. 1986;239:405–409.
- Hagihara M, Nishigaki I, Maseki M, Yagi K. Age-dependent changes in lipid peroxide levels in the lipoprotein fractions of human serum. J Gerontol. 1984;39:269–272.
- Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD, et al. 4- Hydroxynonenal, an aldehydic product of membrane lipid peroxidation, impairs glutamate transport and mitochondrial function in synaptosomes. Neuroscience. 1997;806:85–96.
- Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. Activation of stress signaling pathways by the end product of lipid peroxidation. 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. J Biol Chem. 1999;274:2234–2242.
-
Suc I, Meilhac O, Lajoie-Mazenc I, Vandaele J, Jurgens G, Salvayre R, Negre-Salvayre A. Activation of EGF receptor by oxidized LDL. FASEB J. 1998;12:665–671.
-
Tsukagoshi H, Kawata T, Shimizu Y, Ishizuka T, Dobashi K, Mori M. 4-Hydroxy-2-nonenal enhances fibronectin production by IMR-90 human lung fibroblasts partly via activation of epidermal growth factor receptor-linked extracellular signal-regulated kinase p44/42 pathway. Toxicol Appl Pharmacol. 2002;184:127–135.
- Montuschi P, Collins JV, Ciabattoni G, Lazzeri N, Corradi M, Kharitonov SA, Barnes PJ. Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am J Respir Crit Care Med. 2000;162:1175–1177.
- Morrison D, Rahman I, Lannan S, MacNee W. Epithelial permeability, inflammation, and oxidant stress in the air spaces of smokers. Am J Respir Crit Care Med. 1999;159:473–479.
- Nowak D, Kasielski M, Antczak A, Pietras T, Bialasiewicz P. Increased content of thiobarbituric acid-reactive substances and hydrogen peroxide in the expired breath condensate of patients with stable chronic obstructive pulmonary disease: no significant effect of cigarette smoking. Respir Med. 1999;93:389–396.
- Kelly FJ, Mudway IS. Protein oxidation at the air-lung interface. Amino Acids. 2003;25:375–396.
- Dean RT, Roberts CR, Jessup W. Fragmentation of extracellular and intracellular polypeptides by free radicals. Prog Clin Biol Res. 1985;180:341–350.
- Keck RG. The use of t-butyl hydroperoxide as a probe for methionine oxidation in proteins. Anal Biochem. 1996;236:56–62.
- Davies KJ. Protein damage and degradation by oxygen radicals. I. General aspects. J Biol Chem. 1987;262:9895–9901.
- Stadtman ER. Metal ion-catalyzed oxidation of proteins: biochemical mechanism and biological consequences. Free Radic Biol Med.
1990;9:315–325. - Fucci L, Oliver CN, Coon MJ, Stadtman ER. Inactivation of key metabolic enzymes by mixed-function oxidation reactions: possible implication in protein turnover and ageing. Proc Natl Acad Sci U S A. 1983;80:1521–1525.
- Stadtman ER, Moskovitz J, Levine RL. Oxidation of methionine residues of proteins: biological consequences. Antioxid Redox Signal. 2003;5:577–582.
- Stadtman ER, Levine RL. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids. 2003;25:207–218.
- Stadtman ER. Protein oxidation in aging and age-related diseases. Ann N Y Acad Sci. 2001;928:22–38.
- Shacter E. Quantification and significance of protein oxidation in biological samples. Drug Metab Rev. 2000;32:307–326.
- Poli G, Leonarduzzi G, Biasi F, Chiarpotto E. Oxidative stress and cell signalling. Curr Med Chem. 2004;11:1163–1182.
- Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22.
- Sundaresan M, Yu ZX, Ferrans VJ, Sulciner DJ, Gutkind JS, et al. Regulation of reactive-oxygen species generation in fibroblasts by Rac1. Biochem J. 1996;318:379–382.
- Sun T, Oberley LW. Redox regulation of transcriptional activators. Free Radic Biol Med. 1996;21:335–348.
- Klatt P, Molina EP, De Lacoba MG, Padilla CA, Martinez-Galesteo E, Barcena JA, Lamas S. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J. 1999;13:1481–1490.
- Reynaert NL, Ckless K, Guala AS, Wouters EF, van der Vliet A, Janssen Heininger
YM. In situ detection of S-glutathionylated proteins following glutaredoxin-1 catalyzed cysteine derivatization. Biochim Biophys Acta. 2006;1760:380–387. - Reynaert NL, Wouters EF, Janssen-Heininger YM. Modulation of glutaredoxin-1
expression in a mouse model of allergic airway disease. Am J Respir Cell Mol Biol. 2007;36:147–151. - Filomeni G, Rotilio G, Ciriolo MR. Cell signalling and the glutathione redox system. Biochem Pharmacol. 2002;64:1057–1064.
- Pande V, Ramos MJ. Molecular recognition of 15-deoxydelta (12,14) prostaglandin J(2) by nuclear factor-kappa B and other cellular proteins. Bioorg Med Chem Lett. 2005;15:4057–4063.
- Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62.
- Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–6684.
- Hirota K, Murata M, Sachi Y, Nakamura H, Takeuchi J, Mori K, Yodoi J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J Biol Chem. 1999;274:27891–27897.
- Ward PA. Role of complement, chemokines and regulatory cytokines in acute lung injury. Ann N Y Acad Sci. 1996;796:104–112.
- Akira S, Kishimoto A. NF-IL6 and NF-kB in cytokine gene regulation. Adv Immunol. 1997;65:1–46.
- Meyer M, Schreck R, Baeuerle PA. H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. EMBO J. 1993;12:2005–2015.
- Abate C, Patel L, Rausher FJ, Curran T. Redox regulation of fos and jun DNA-binding activity in vitro. Science. 1990;249:1157–1161.
- Galter D, Mihm S, Droge W. Distinct effects of glutathione disulphide on the nuclear transcription factors kB and the activator protein-1. Eur J Biochem. 1994;221:639–648.
- Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc Natl Acad Sci U S A. 1997;94: 3633–3638.
Post Disclaimer
Professional Scope of Practice *
The information herein on "El Paso, TX Oxidative Stress and Antioxidant Defense" is not intended to replace a one-on-one relationship with a qualified health care professional or licensed physician and is not medical advice. We encourage you to make healthcare decisions based on your research and partnership with a qualified healthcare professional.
Blog Information & Scope Discussions
Welcome to El Paso's Premier Wellness, Personal Injury Care Clinic & Wellness Blog, where Dr. Alex Jimenez, DC, FNP-C, a Multi-State board-certified Family Practice Nurse Practitioner (FNP-BC) and Chiropractor (DC), presents insights on how our multidisciplinary team is dedicated to holistic healing and personalized care. Our practice aligns with evidence-based treatment protocols inspired by integrative medicine principles, similar to those on this site and our family practice-based chiromed.com site, and focuses on restoring health naturally for patients of all ages.
Our areas of multidisciplinary practice include Wellness & Nutrition, Chronic Pain, Personal Injury, Auto Accident Care, Work Injuries, Back Injury, Low Back Pain, Neck Pain, Migraine Headaches, Sports Injuries, Severe Sciatica, Scoliosis, Complex Herniated Discs, Fibromyalgia, Chronic Pain, Complex Injuries, Stress Management, Functional Medicine Treatments, and in-scope care protocols.
Our information scope is multidisciplinary, focusing on musculoskeletal and physical medicine, wellness, contributing etiological viscerosomatic disturbances within clinical presentations, associated somato-visceral reflex clinical dynamics, subluxation complexes, sensitive health issues, and functional medicine articles, topics, and discussions.
We provide and present clinical collaboration with specialists from various disciplines. Each specialist is governed by their professional scope of practice and their jurisdiction of licensure. We use functional health & wellness protocols to treat and support care for musculoskeletal injuries or disorders.
Our videos, posts, topics, and insights address clinical matters and issues that are directly or indirectly related to our clinical scope of practice.
Our office has made a reasonable effort to provide supportive citations and has identified relevant research studies that support our posts. We provide copies of supporting research studies upon request to regulatory boards and the public.
We understand that we cover matters that require an additional explanation of how they may assist in a particular care plan or treatment protocol; therefore, to discuss the subject matter above further, please feel free to ask Dr. Alex Jimenez, DC, APRN, FNP-BC, or contact us at 915-850-0900.
We are here to help you and your family.
Blessings
Dr. Alex Jimenez DC, MSACP, APRN, FNP-BC*, CCST, IFMCP, CFMP, ATN
email: [email protected]
Multidisciplinary Licensing & Board Certifications:
Licensed as a Doctor of Chiropractic (DC) in Texas & New Mexico*
Texas DC License #: TX5807, Verified: TX5807
New Mexico DC License #: NM-DC2182, Verified: NM-DC2182
Multi-State Advanced Practice Registered Nurse (APRN*) in Texas & Multi-States
Multi-state Compact APRN License by Endorsement (42 States)
Texas APRN License #: 1191402, Verified: 1191402 *
Florida APRN License #: 11043890, Verified: APRN11043890 *
Colorado License #: C-APN.0105610-C-NP, Verified: C-APN.0105610-C-NP
New York License #: N25929, Verified N25929
License Verification Link: Nursys License Verifier
* Prescriptive Authority Authorized
ANCC FNP-BC: Board Certified Nurse Practitioner*
Compact Status: Multi-State License: Authorized to Practice in 40 States*
Graduate with Honors: ICHS: MSN-FNP (Family Nurse Practitioner Program)
Degree Granted. Master's in Family Practice MSN Diploma (Cum Laude)
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)
(Licensed Medical Doctor)
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933
Licenses and Board Certifications:
MD: Medical Doctor
DC: Doctor of Chiropractic
APRNP: Advanced Practice Registered Nurse
FNP-BC: Family Practice Specialization (Multi-State Board Certified)
RN: Registered Nurse (Multi-State Compact License)
CFMP: Certified Functional Medicine Provider
MSN-FNP: Master of Science in Family Practice Medicine
MSACP: Master of Science in Advanced Clinical Practice
IFMCP: Institute of Functional Medicine
CCST: Certified Chiropractic Spinal Trauma
ATN: Advanced Translational Neutrogenomics
Memberships & Associations:
TCA: Texas Chiropractic Association: Member ID: 104311
AANP: American Association of Nurse Practitioners: Member ID: 2198960
ANA: American Nurse Association: Member ID: 06458222 (District TX01)
TNA: Texas Nurse Association: Member ID: 06458222
NPI: 1205907805
| Primary Taxonomy | Selected Taxonomy | State | License Number |
|---|---|---|---|
| No | 111N00000X - Chiropractor | NM | DC2182 |
| Yes | 111N00000X - Chiropractor | TX | DC5807 |
| Yes | 363LF0000X - Nurse Practitioner - Family | TX | 1191402 |
| Yes | 363LF0000X - Nurse Practitioner - Family | FL | 11043890 |
| Yes | 363LF0000X - Nurse Practitioner - Family | CO | C-APN.0105610-C-NP |
| Yes | 363LF0000X - Nurse Practitioner - Family | NY | N25929 |
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)*
(Licensed Medical Doctor)*
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933


 Again, We Welcome You.
Again, We Welcome You.
Comments are closed.